Development process: Lean and compliant with standards
Laws and standards formulate requirements on how medical device manufacturers must define and document development processes. Notified bodies check these requirements during audits.
This article on development processes provides tips on how to design the processes and how to align it with other processes, such as the risk management process.
1. Development process: Basics
a) Definition
Development processes define how the roles involved in the development perform which activities and in which order in order to transform given inputs into outputs.
Elements to be determined | Examples |
Activities (what?) | Verify the system requirements Define the architecture Derive test cases Perform tests |
Order of activities (when?) | Sequential (waterfall model), parallel or iterative |
Methods and procedures (how?) | Inspection with checklist to review a document Modeling with UML to describe an architecture Black box test procedure for the systematic derivation of test cases |
Roles involved (who?) | Head of engineering Software architect Software developer Tester |
Inputs | Stakeholder requirements Organizational requirements |
Outputs | Construction plans (e.g., architecture) Software code Test results |
Tab. 1: Elements that a development process must determine
b) Objectives
Defining a development process is part of the constructive quality assurance, i.e., quality assurance with the objective of avoiding errors.
From a regulatory point of view, the process for medical devices should ensure that the devices have the specified safety, performance, and effectiveness.
Furthermore, the process should contribute to efficiency:
- For example, resources are used in the best possible way.
- The people involved in the development process are coordinated in the best possible way.
- The development project is completed in the planned time.
- Last but not least, development risks are identified at an early stage.
2. Regulatory requirements for the development process
Most regulations and standards follow a process-oriented approach. They therefore impose requirements on the processes.
Regulatory documents | Requirements for the processes |
The QM system describes the processes and procedures. The software-lifecycle processes must be defined. | |
The standard requires the establishment of five processes, including a development process. | |
The standard follows a process-oriented concept. It requires a total of 25 procedures, among others for development. | |
The standard requires a risk management process. This should be coordinated with the development process (see chapter 4). | |
The standard requires a usability engineering process. This should be coordinated with or integrated into the development process (see chapter 5). | |
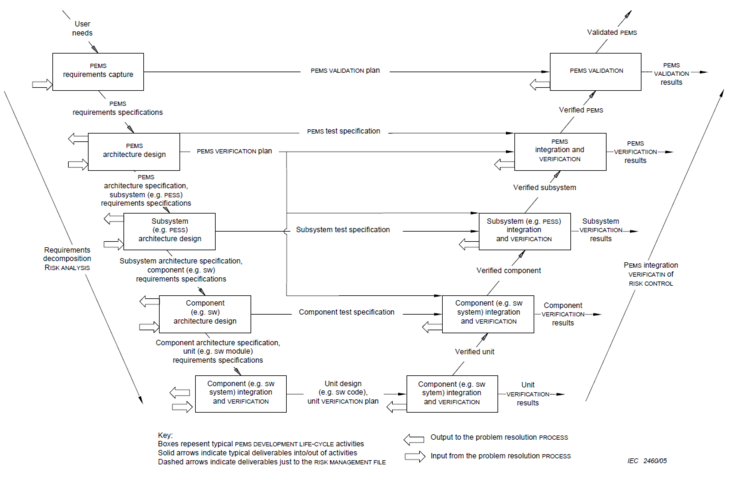
At least for programmable electrical medical systems, the standard insists on a development process. In the appendix, it shows a process model similar to the V-model (see Fig. 1). |
Tab. 2: Regulatory requirements for the development process
None(!) of the regulations requires a specific process model.
Read below how you can avoid redundant efforts and problems in the audit by aligning the development process and the risk management and usability engineering process.
3. Practical tips for the development process
Tip 1: Use the V-model as a documentation model
Most companies work according to a waterfall, V-model, or agile process model.
Further information
Read more about agile software development.
No matter which process model you decide on, you must
1. commit to one model (at least per project) and document your decision,
2. work according to this process model and thereby
3. perform and document the activities required by the regulations.
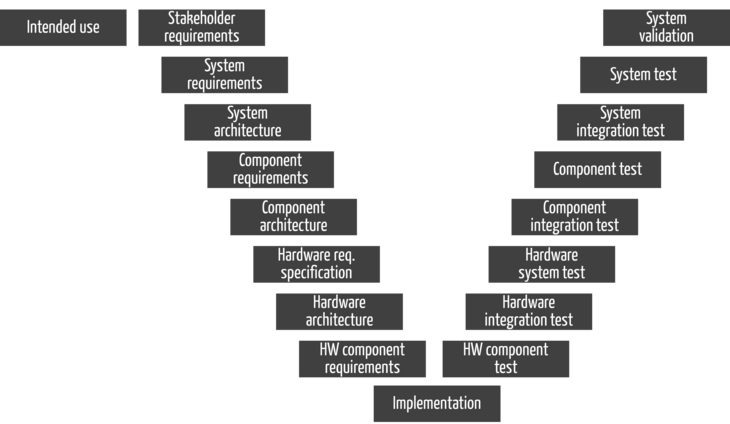
These activities must result in documents as they would in the V-model (see Fig. 2). However, this does not mean that manufacturers must develop according to the V-model.
Be sure to note!
Note that you complete all activities on the left side in Fig. 2 (i.e., all documents) with a verification before releasing them.
Tip 2: Benefit from agile development process and avoid pitfalls
In practice, iterative-incremental approaches prove their worth. This is because it is usually not possible to achieve the outputs of a phase / activity completely and correctly right away. This is evident, for example, when eliciting stakeholder requirements and creating the architecture. For example, it is usually not possible to determine the time behavior and resource consumption (e.g., CPU) without experimentation.
However, even with an iterative and incremental approach, the manufacturer must not refrain from verifying the outputs of a previous activity before starting the next one. Nor is it efficient to replace systematic requirements engineering with a try-and-error approach.
Read more about the pitfalls in this article on agile (software) development.
Tip 3: Scoping the development process correctly
Fig. 2 suggests that the development process covers all activities. While the manufacturer must cover all activities through a process; it makes perfect sense that the development team is only responsible for the activities that come after the elicitation and validation of stakeholder requirements.
In addition, the development team can limit the description of the development process to its area of responsibility (see Fig. 3). Then the elicitation of the requirements would be regulated by another process.
Tip 4: Separate research and development
It is helpful to free the research team as much as possible from regulatory constraints. After all, its objective should also be to minimize development risks by testing (researching) solutions and technologies.
4. Interaction with the risk management process
a) Problem definition
Most standards relevant to medical device development, such as ISO 13485, ISO 14971, IEC 62304, and IEC 62366-1, impose requirements on processes.
To meet these requirements, many medical device manufacturers formulate process and procedure descriptions (SOPs) - separately for each of these processes. As a result, those involved in development must follow several of these SOPs at the same time. Errors are then almost inevitable.
b) Solution
Although the following figure shows a V-model, the considerations are independent of it.
Overview
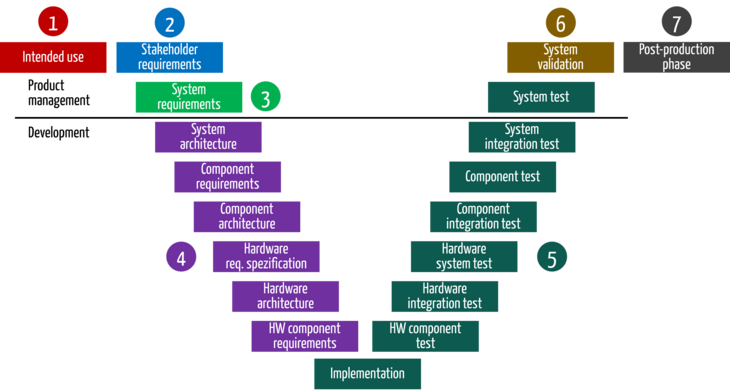
The description of the development process should include references to the relevant activities in the risk management process at each stage. This ensures that risk management is not forgotten.
Individual phases
| Phase in the development process | Activities in the development process | Activities in the risk management process |
1 | Context and business model | Define the business expectations and context for the device | Start the risk analysis with a preliminary hazard analysis. It is already obvious from the intended purpose / use which hazards and risks the device causes. |
2 | Stakeholder requirements | Identify stakeholder requirements Determine state of the art | Determine risk policy and the risk acceptance criteria |
3 | System specification | Derive system requirements and system specifications. In doing so, define requirements for the device from a black box view and thus its inputs and outputs. | The input and output analysis helps to identify further hazards and risks. With this part of the risk analysis, manufacturers should be able to complete the preliminary hazard analysis and, thus, the identification and assessment of the most important hazards or risks. |
4 | System design | Design product or system, e.g., create architecture, create software design, design isolation diagrams | The risk analysis can be completed when requirements have been transformed into a solution. The way the device is to be built introduces additional risks or helps to minimize them. Risks can be identified with an FMEA. At the same time, planning of the measures that minimize the risk and with which the functional safety of the device must be achieved begins at this stage at the latest. |
5 | Implementation and verification | Programming software Build first devices (prototypically) Verify both with tests, e.g., software system test | Verify effectiveness of risk mitigation measures |
6 | Validation | Validate device, e.g., with summative evaluation (usability tests) and clinical evaluation | The purpose of validation is to demonstrate not only the safety of the devices, but also their benefits, and thus that the risk-benefit ratio is acceptable. |
7 | Production and post-production phase | No longer subject to development | Identifying risks during production, shipping and putting into service As part of post-market surveillance, ongoing inspection to ensure that all risks are fully and correctly assessed and are still acceptable in comparison to the benefit |
Tab. 3: "Mapping" of activities in the development process and risk management process
5. Interaction with the usability engineering process
Quality management and usability engineering are closely interrelated as well: While ISO 13485 formulates general concepts and requirements, IEC 62366-1 provides concrete requirements in the context of usability. This article shows the mapping of both standards.
Quality Management ISO 13485 | Usability |
Chapter 6.1: Provision of resources | 4.3 Adjusting the usability engineering effort |
Chapter 6.2: Human resources | 4.1.1 Usability engineering development process |
Chapter 7.1: Product realization planning | 4.3 Adjusting the usability engineering effort |
Chapter 7.3.2 Development planning | 5.5 Selecting the hazard-related use scenarios for the summative evaluation 5.7 Creating a plan for the user interface evaluation |
Chapter 7.3.3 Development inputs | 5.1 Creating the use specification 5.2 Determine characteristics of the user interface with respect to safety and possible use errors 5.3 Identify known or foreseeable hazards and hazardous situations. 5.4 Identify and describe hazard-related use scenarios. 5.6 Create the user interface specification |
Chapter 7.3.4 Development outputs | 5.8 Performing user interface design, implementation, and formative evaluation |
Chapter 7.3.5 Development review | 5.8 Performing user interface design, implementation, and formative evaluation |
Chapter 7.3.6 Development verification | 5.8 Performing user interface design, implementation, and formative evaluation |
Chapter 7.3.7 Development validation | 5.8 Performing user interface design, implementation, and formative evaluation |
Tab. 4: Interaction of the processes according to ISO 13485 and IEC 62366-1
6. Conclusion and summary
Manufacturers should spend enough energy to describe their development process(es) precisely and specifically. Because otherwise, problems are imminent.
Mistake | Possible consequences |
In the worst case, the process is not described at all or not described completely. | This leads to non-conformity (notified bodies react sensitively when processes are not defined and lived). There is a risk of chaos in development |
The process is described inaccurately. | Regulatory requirements are not met, and discrepancies loom in audits. The team does not know how to proceed, resulting in unsafe devices and delayed projects. |
The process is described accurately but inappropriately. | The team (rightly) perceives the process as bureaucratic and obstructive. The team either deviates from the process (non-conformance) or it works inefficiently and ineffectively. |
The effort pays off: With a good development process, development projects lead quickly and predictably to safe devices, satisfied customers and economic success.

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.