Audit: Prerequisite for ISO 13485 Certification
In most cases, an audit by a notified body is a prerequisite to market medical devices in the European market.
Definitions
Definition: Audit
„Systematic, independent and documented process for obtaining audit evidence and evaluating it objectively to determine the extent to which the audit criteria are fulfilled“
Source: ISO 9000:2015
Auditing Medical Device Manufacturers
Europe
As for medical device manufacturers, the ISO 13485 audit is the most important one. Only if this audit has been passed successfully, notified bodies may issue an ISO 13485 respectively annex II certificate. Those certifications, in return, are requirements for a conformity assessment according to annex II of the Medical Device Directive (MDD) respectively annex IX of the Medical Device Regulation (MDR) and thus for legally marketing the products.
ISO 13485 Audits may only be performed by accredited bodies. When interpreting ISO 13485, auditors rely on the explanation of ISO 14969.
USA / FDA
The FDA does not perform audits, but rather inspections. This means that at best, inspections by the FDA are concluded without any findings but do not lead to the issue of a certificate. In contrast, audits by European notified bodies, at best, result in the issue of a certificate. The FDA does not check compliance with ISO 13485, but with 21 CFR part 820.
Audits: Different Types
Depending on the audit's focus, the following types can be distinguished:
- system audit (e.g. conformity testing of a quality management system with requirements laid out in a standard such as ISO 13485 or ISO 9001)
- process audit
- product audit
- software audit
Further distinctions are:
- supplier audits (can, again, take the form of system or process audit)
- unannounced audits
- internal audits
Demands on Audits and Auditors
ISO 19011 describes demands on audits (planning, execution and documentation) and auditors.
Further provisions stipulate the duration of audits.
FAQ: Answers to most frequent questions
- When must audits be carried out? An audit by a notified body is required if you intend to place a medical device on the European market. Products classified as class I are an exemption.
- How long does an audit take? This, inter alia, depends on the size of your company. As a rule of thumb companies with 5 employees have to expect 2,5 days, with 10 employees 3 days, with 50 employees 6 days and with 100 employees 8 days.
- Who may perform audits? This depends on the audit's objective. If you aim at a certification pursuant to ISO 13485, notified bodies are exclusively authorized.
- Which requirements must be met in context of audits? Regarding an ISO 13485 audit, you must have and follow a documented quality management system. This, for example, means that you must have at least for the most part developed and produced a product compliant to the standard's requirements. Hence, a medical device's technical documentation must be (substantially) complete. Auditors would like to examine if your QM-system meets the normative demands and if you abide by your QM-system.
- Support: Who can support you in preparing for an audit? The Johner Institute supports you in various ways:
- Complimentary Consulting: answering session with our team, often even free of charge.
- The training videos explain how to create a lawful documentation, step by step.
- The consulting team supports you in writing a QM handbook and establishing a QM-system compliant with standards.
- As part of mock-audits, the auditors of the Johner Institute examine if your QM-system is ready for an audit by a notified body.
- In seminars at the Johner Institute, you will learn about how to meet all regulatory requirements.
Get in contact with us to find out how to quickly establish a lean and regulatory compliant QM system and thus to lay the foundations for bringing your medical devices to market.
What Auditors Assess
If the auditor attests "non-conformity", your notified body may refuse or withdraw the certificate. Withal it is known in general what the auditor examines:
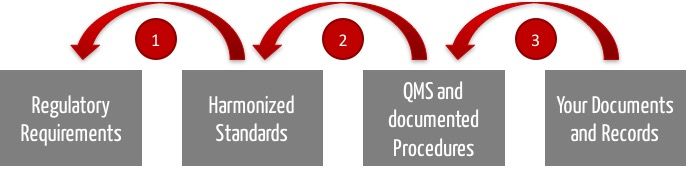
The following numbers refer to the above figure.
- The auditor must ensure that you act compliant with the law. According to the Medical Device Directive / Regulation and thus to medical device law, he can/shall/may presume that your product development is lawful if you comply with the respective harmonized standards. Among them are, inter alia, ISO 13485 concerning QM-system, ISO 14971 regarding risk management, IEC 62304 on software development processes and IEC 62366 covering the issue of usability.
- Thus, the audit will examine if your QM-system - i.e. the rules you are imposing on yourself - complies with the standard. For example, if your QM system covers all aspects of the ISO 13485 standard. In general, the auditor even announces which aspects (chapters of the standard) he will primarily examine.
- Finally, your auditor will check if you have been playing by your own rules. If you have done exactly the things your QM-system requires you to do when developing specific products and if you have kept records.
If there are deviations regarding the 2nd and 3rd point, you will have a problem.
E-Learning Platform
The training videos show you how to compile a documentation compliant with FDA and standards with which you will shine in audits.
View and sign-up for the training videos

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.