MDCG 2020-5: The End of the Equivalence Route for Clinical Evaluations?
The document MDCG 2020-5 (“Clinical Evaluation – Equivalence”) increases the requirements for the equivalence of medical devices that manufacturers can refer to in the clinical evaluation of their device.
A clear understanding of the regulatory requirements will help you:
- Quickly decide whether you really want to down the equivalence route or you would rather use your own clinical data and/or performance data
- Robustly demonstrate the equivalence of other devices to ensure that your clinical evaluation satisfies your notified body
This will help you avoid unnecessary iterations and costs and create the conditions required for a quick authorization of your device.
1. Regulatory requirements for equivalence
a) The MDR defines the requirements for the equivalence of devices
Aspects of equivalence
In Annex XIV Part A (3), the MDRdefines the conditions to be met for a device to be regarded as equivalent to a manufacturer’s own device in the clinical evaluation: A device is only equivalent if
- technically
- biologically and
- clinically
it has the same characteristics. It is precisely this equivalence that manufacturers have to demonstrate in the clinical evaluation. Otherwise, they cannot use the clinical data from the other device to demonstrate the safety, performance and clinical benefit of their own device.
This requirement is included in the definition of the term “clinical data”:
‘clinical data’ means information concerning safety or performance that is generated from the use of a device and is sourced from the following: […] reports published in peer reviewed scientific literature on other clinical experience of either the device in question or a device for which equivalence to the device in question can be demonstrated,
Source: MDR Article 2
The second section describes the MDR's requirements for technical, biological and medical equivalence in more detail.
Further information
Read more on clinical data and clinical evaluations.
Proving equivalence requires access to data
The manufacturer has to have data that support the claims in the clinical evaluation.
“It shall be clearly demonstrated that manufacturers have sufficient levels of access to the data relating to devices with which they are claiming equivalence in order to justify their claims of equivalence.”
Source: MDR Annex XIV Part A (3)
Special requirements for specific device classes
The MDRestablishes additional requirements for specific device classes. These limit the number of manufacturers and devices that can be included in a clinical evaluation based on the equivalence route (i.e. without a clinical investigation):
Device class | Manufacturer | Explanation |
Class IIb implantable and class III | Same | Predecessor device with modifications with CE-marked predecessor device |
Class IIb implantable and class III | Different | Full access to technical documentation regulated by contract |
Device without intended medical purpose | Not important | Justification when existing clinical data from an analogous medical device is used |
Other devices | Not important | Access to pre-clinical and clinical data |
b) MEDDEV 2.7/1 rev. 4 clarifies the equivalence requirements
Revision four of the MEDDEV 2.7/1 guideline contains a two-page annex, “A1”, that specifies more precisely the conditions under which clinical, technical and biological equivalence may be assumed to exist.
Section 2 below gives a more detailed overview of these requirements.
The MEDDEV document contains additional requirements.
Further information
Read more on the topic of MEDDEV 2.7/1 Revision 4.
Equivalence can only be demonstrated on the basis of a single medical device
Manufacturers may be tempted to use a different comparator device for each aspect of equivalence (technical, biological, clinical – see Fig. 1). This is exactly what MEDDEV 2.7/1 rules out.
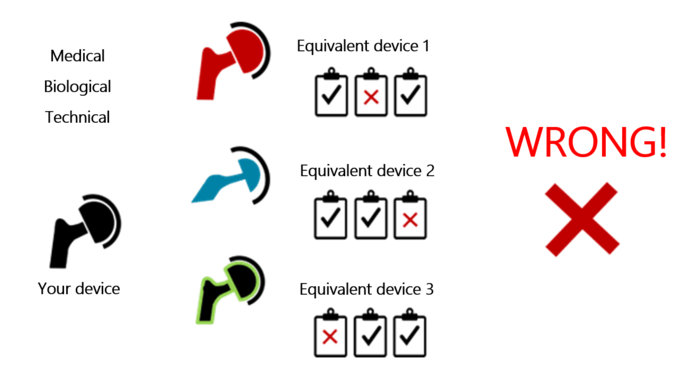
Manufacturers can only use a device’s clinical data if the device is equivalent in all three aspects (see Fig. 2). Otherwise, it does not count as an equivalent device.
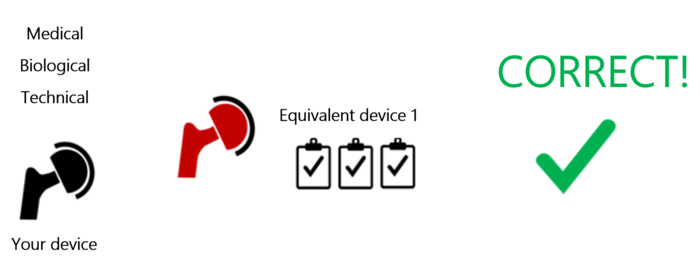
However, manufacturers can use data from more than one equivalent device if they are all equivalent in all three aspects.
The manufacturer also has to disclose the differences between their device and the comparator device
MEDDEV also requires manufacturers to disclose all the differences between their own device and the device they are claiming is an equivalent device. The guideline expects explanations why these differences do not significantly affect clinical performance and the clinical safety.
The equivalent device must be CE-marked
The requirement that only clinical data from CE-marked medical devices can be used in clinical evaluations has led to a lot of discussion.
When reviewing the technical documentation, notified bodies generally no longer insist on this requirement and also accept, for example, devices that have been authorized in the USA as equivalent devices.
c) MDCG 2020-5 “Clinical Evaluation – Equivalence”
The MDCG document is actually aimed at notified bodies. However, because it explains the requirements of the MDR and MEDDEV 2.7/1, manufacturers should also take a look at this MDCG guideline.
The MDCG guideline discusses the differences between the MDR and MEDDEV 2.7/1
The requirements of the MDR and MEDDEV are not exactly the same. In some cases, the MEDDEV requirements go beyond those of the MDR, and in other cases the MDR contains requirements that the MEDDEV document does not.
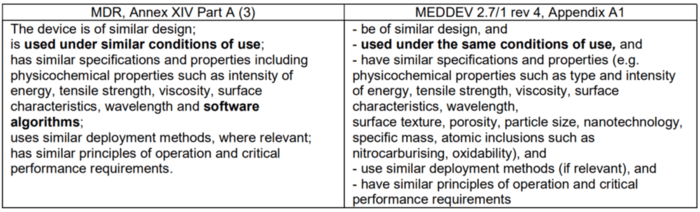
For example, if the devices are technically equivalent, the MDR requires the software algorithms be considered. The MEDDEV document does not address this. The MDCG-2020-5 guideline repeatedly emphasizes the relevance of these software algorithms, unless the software does not have a medical purpose but, for example, only controls the hardware.
According to MDCG 2020-5, the requirement for the software algorithms to be equivalent does not extend to the equivalence of the software code itself. “Only” the functional principle, the clinical performance(s) and the intended purpose(s) of the software algorithm are relevant.
However, the guideline insists that the code must have been developed in line with international standards, such as IEC 62304. Whether this is referring to the code of the manufacturer’s device or of the equivalent device is not clear. If it is the manufacturer's device, this would be a regulatory requirement in any case.
MDCG 2020-5 warns against confusing “similar” and “equivalent”
A lot of manufacturers confuse equivalent and similar devices when preparing their clinical evaluation. The MDCG 2020-5 guideline warns against precisely that. The MDCG defines similar devices as follows:
“The term ‘similar devices’ may be understood as devices belonging to the same generic device group. The MDR defines this as a set of devices having the same or similar intended purposes or a commonality of technology allowing them to be classified in a generic manner not reflecting specific characteristics.”
Source: MDCG 2020-5, section 5
As similar devices are not compared on the basis of specific characteristics, manufacturers cannot use them for the equivalence assessment.
However, data from similar devices can be useful for a variety of other purposes. The “Tips” section provides further information.
MDCG 2020-5 specifies the requirements for scientific evidence
The MDCG 2020-5 document wants manufacturers to document the comparisons for technical, biological and clinical equivalence in a table. As a result, the guideline provides tables containing the attributes to be compared (see Fig. 4). The second section looks at these attributes (characteristics).
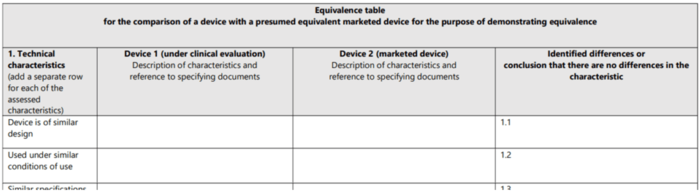
MDCG 2020-05 expects evidence for each characteristic. This evidence must be based on valid scientific data, such as:
- Clinical data from the literature
- Published scientific data from animal studies
- Pre-clinical data from the manufacturer's technical documentation, such as
- Specifications
- Test results
- Chemical/physical/biological analyses
- Data from pre-clinical investigations etc.
These pre-clinical and clinical data from the equivalent device must relate to a defined generation or version of the device.
2. Demonstrating equivalence in accordance with the MDR, MEDDEV 2.7/1 and MDCG 2020-5
a) Clinical equivalence
The MDR defines the criteria for clinical equivalence:
“The device is used for the same clinical condition or purpose, including similar severity and stage of disease, at the same site in the body, in a similar population, including as regards age, anatomy and physiology; has the same kind of user; has similar relevant critical performance in view of the expected clinical effect for a specific intended purpose.”
Source: MDR Annex XIV Part A (3)
That means that your own device and the equivalent device must be used:
- under the same clinical conditions for the same medical purpose
- for the same patients (including stage of disease, anatomy and physiology) and
- by the same users
Otherwise, the “equivalence route” cannot be used for the clinical evaluation.
Comparing these attributes in a table is recommended:
Clinical characteristics | Own device | Equivalent device | Evaluation |
Clinical condition or purpose |
|
| Same |
Patient population: Age | 18 years and older | Adults | Same |
Patient population: Disease (severity, stage) | [..] | [..] | [..] |
Users | Medical professionals | Physicians, intensive care staff | Same |
Performance in terms of expected clinical effect | [..] | [..] | Similar |
User equivalence
According to the MDCG-5 document, a user means any healthcare professional or lay person who uses a device.
A lay person can be an individual who does not have formal education in a relevant field of healthcare or a medical discipline.
You must therefore take into consideration whether the intended user’s competence or knowledge could have an effect on the safety, clinical performance and desired clinical outcome.
Clinical conditions, clinical purpose, patient population
The equivalent device should be intended for use under the same clinical conditions or for the same purpose, including for similar severities and stages of disease.
The MDR does not explicitly require the same patient population, but it does require a scientific justification as to why relevant aspects do not play a significant role with regard to clinical performance, safety and benefits of the device.
For this, the following aspects should be considered in the clinical evaluation:
- Age
- Gender
- Physiology
- Anatomy
- And any other relevant aspects
However, the devices must be used at the same site in the body.
The equivalent device must also have a similar relevant critical performance in view of the expected clinical effect for a specific intended have purpose.
b) Biological equivalence
The MDR defines the criteria for biological equivalence:
“The device uses the same materials or substances in contact with the same human tissues or body fluids for a similar kind and duration of contact and similar release characteristics of substances, including degradation products and leachables;”
Source: MDR Annex XIV Part A (3)
This means that the equivalence route is only possible if your device and the equivalent device are made of the same materials or substances in contact with the same human tissues or body fluids. Otherwise, this is the end of the equivalence route.
Tables are also recommended here to compare the attributes:
Biological characteristics | Own device | Equivalent device | Evaluation |
Materials and substances | A, B and C | B, C and D | Not the same (end) |
Contact with human tissue or body fluids | A and B, not C | C | Not the same |
Type and duration of contact | 2 h, direct skin contact | Undefined, direct skin contact | Similar |
Release characteristics of substances, degradation products, leachables | […] | […] | […] |
The distinction between the same materials or substances and similar release characteristics of substances is only made because the processing, design and use environment can influence the release characteristics of the material. Even when the raw materials are the same.
Processing can also make materials more susceptible to degradation. For example, small changes in pH or oxidative stress can increase or decrease such release. For this reason, it is the final device that should always be tested.
The equivalence comparison is even more demanding for devices made of substances or combinations of substances or even ones that contain medicinal products.
“Substances” and “combinations of substances” also refer to all associated ancillary substances and coatings. In this case, manufacturers have to review additional characteristics for the clinical evaluation, such as:
- Absorption in the body
- Distribution in the body
- Metabolism
- Excretion
- Local tolerance
- Toxicity
- Interaction with other devices, medicinal products or other substances and the potential for adverse reactions.
- The benefit of the substance
The MDCG guideline refers to the ISO 10993 series of standards.
Further information
Read more on biocompatibility and ISO 10993.
MEDDEV 2.7/1 allows manufacturers to make exceptions if their own device and the comparator device do not use the same substances or materials. The MDCG 2020-05 guideline points out that these exemptions are no longer permitted under the MDR.
c) Technical equivalence
According to the MDR, the technical equivalence only has to be a similarity, but there are limits here as well.
“The device is of similar design; is used under similar conditions of use; has similar specifications and properties including physicochemical properties such as intensity of energy, tensile strength, viscosity, surface characteristics, wavelength and software algorithms; uses similar deployment methods, where relevant; has similar principles of operation and critical performance requirements;”
Source: MDR Annex XIV Part A (3)
An equivalence table should, for example, compare the following attributes of the two devices:
Technical characteristics | Own device | Equivalent device | Evaluation | ||
Design |
|
|
| ||
Conditions of use |
|
|
| ||
Specifications and physicochemical properties |
|
|
| ||
Deployment methods |
|
|
| ||
Principles of operation |
|
|
| ||
The conditions of use must be similar to the extent that there are no clinically significant differences in safety and clinical performance between your device and the equivalent device. The conditions of use include the physical environment (e.g., brightness, moisture, vibrations, contamination, temperature and pressure).
For the specifications and physicochemical properties, manufacturers should consider the following:
- Type and intensity of energy
- Tensile strength
- Viscosity
- Surface characteristics
- Wavelength
- Surface texture
- Porosity
- Particle size
- Nanotechnology
- Specific mass
- Atomic inclusions such as nitrocarburizing, oxidability
The software is considered one of the principles of operation. We already looked at this aspect in section 1.c).
Technical equivalence only requires similarity for a lot of characteristics. However, these differences must be scientifically justified.
3. Tips
a) Add missing data
Manufacturers rarely have access to the technical document for competitors’ devices. If data required to demonstrate equivalence for some attributes is missing, it may be helpful to generate this data through comparative testing.
If these tests also fail to provide sufficient evidence or conformity, a clinical investigation becomes more likely. Manufacturers should then be careful to specify what evidence they want to use. The endpoints of the clinical investigation should be defined and the study protocols designed with this evidence in mind.
b) You can still use data from similar devices
Manufacturers cannot use (clinical) data from devices that are similar but not equivalent to demonstrate safety, performance and clinical efficacy. But this data can still be useful for a variety of other purposes:
- Ensuring that the risk management system is comprehensive
- Understanding the state of the art in the clinical evaluation
- To help define the scope of the clinical evaluation by identifying design features in similar devices that raise particular concerns.
- To help plan
- clinical investigations
- the post-market clinical follow-up
- the post-market surveillance
4. Summary
a) Advantages
The MDCG 2020-5 guideline is aimed at notified bodies. Medical device manufacturers would also be well advised to study this document:
- It will help them to prepare for auditors’ lines of argument.
- The tables help to demonstrate technical, biological and clinical equivalence more systematically.
- The document interprets the differences between the MDR and the MEDDEV 2.7/1 guideline.
- It warns against the common mistake of confusing similar devices with equivalent devices.
b) Quality and consistency of regulatory requirements
However, it is worth thinking about why a document like MDCG 2020-5 is needed:
- the legal requirements are obviously not sufficiently complete or/and understandable.
- Two of the documents issued by the EU are not sufficiently aligned, meaning further explanation is needed.
- There is no systematic and regular maintenance of, for example, MEDDEV 2.7/1, which is why these inconsistencies and contradictions have not been corrected.
- Manufacturers now have to read at least three documents.
- Two of these three documents have no legal force. There are different views on whether they are binding or not.
- The quality assurance processes the MDCG documents go through are still opaque. Every law must go through a consultation procedure and pass a vote in a democratic process. The MDCG guidelines obviously do not.
c) Conclusion
It is clear that the requirements for the equivalence of devices and therefore the clinical evaluation as a whole must be strict. After all, manufacturers have to conclusively demonstrate that their devices are safe, perform as intended and really are beneficial to patients.
But disproportionately high requirements also prevent beneficial devices being authorized and mean avoidable costs for manufacturers. The MDCG has intensified this problem with its 2020-5 document.
The Johner Institute’s team of clinical evaluators has prepared countless clinical evaluations in compliance with the requirements of the MDR, MEDDEV 2.7/1 and MDCG 2020-5 and has safely guided them through review by notified bodies.
Contact us by email or the contact form to get your clinical evaluation reviewed, drafted or improved.

Author:
Dr. Bettina Martin

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.