Technical Documentation for Medical Devices
The term technical documentation (or technical file) refers to the documents that a medical device manufacturer must submit to the authority before placing it on the market. Completing a technical file is an unavoidable step to pass the conformity assessment or approval process. Therefore, it is an important initial undertaking in the quest for the approval of all medical devices.
1. Regulatory requirements for technical documentation
a) Medical Device Directive 93/42/EEC (MDD)
The Medical Device Directive (93/42/EEC) specifies the obligatory requirements for medical devices. Manufacturers are legally obliged to demonstrate compliance with these requirements, through the completion of the technical documentation (also called the TD or technical file).
The Medical Devices Directive (in contrast to the MDR) is somewhat ambiguous with regard to the requirements for these documents. However, it mentions that they must include the following:
- A general description of the product, including any planned variants along with its intended use;
- The design specifications, including the standards which will be applied and the results of the risk analysis, and a description of the solutions adopted to fulfill the essential requirements, which apply, if the standards referred to in Article 5 are not applied in full;
- The techniques used to control and verify the design, the processes, and the systematic measures which will be adopted during the design phase;
- If the device is intended to be connected with another device to operate as intended, proof must be provided that it conforms to the essential requirements when connected to any such device, while upholding the characteristics specified by the manufacturer;
- The solutions adopted in accordance with Annex I Chapter I Section 2;
- The preclinical evaluation;
- The clinical evaluation referred to in Annex X;
- The draft label and, where appropriate, instructions for use.
All above mentioned documents must be submitted by the manufacturer during the conformity assessment procedure via the technical documentation.
b) Medical Device Regulation (MDR)
The Medical Device Regulation (MDR) does not only define the requirements for the medical device (Annex I), but it also defines the requirements for the documentation itself (Annex II). This must include (see Fig. 1):
- Identification of the device (e.g. with a UDI— Unique Device Identifier)
- Description of the device, including variants, configuration and accessories
- Intended use
- Labeling (packaging, instructions for use, etc.)
- Information on the design and manufacture of the device
- A risk management file
- Verification and validation of the device and, therefore, proof that the device meets the general safety and performance requirements
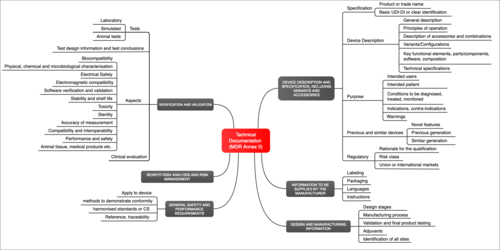
The MDR goes one step further: It includes post-market surveillance, with planning and implementation, under technical documentation. It establishes the corresponding requirements in Annex III.
c) ISO 13485:2016
The 2016 version of ISO 13485 introduced the above mentioned medical device file. This file must provide similar information:
- Description of the device
- Intended purpose
- Labeling (packaging, marking, instructions for use, installation and maintenance instructions)
- Device specification
- Specifications for its manufacture, packaging and storage
- Market surveillance
- Proof of conformity, including verification and validation
d) Notified bodies
The notified bodies have also published recommendations, such as NB-MED/2.5.1.Bear in mind that these publications have no legal standing. However, their contents are regularly requested during audits and reviews of the technical documentation.
e) FDA
The FDA also requires detailed device documentation,comprising three distinct files:
- Design History File (DHF)
- Device Master Record (DMR)
- Device History Record (DHR)
f) Canada
The Canadian authorities have published a unique iteration of the structure of the technical documentation based on the STED format.
2. Contents of technical documentation
The above regulations provide a brief outline of the requirements for technical documentation, yet do not describe:
- Specific questions, e.g. what does software architecture or biocompatibility testing have to describe?
- How the content is organised across the documents
- The chapter structure of the documents
Though standards such as ISO 14971, IEC 62304, IEC 60601-1 and IEC 62366-1, offer some more detailed specifications. ISO 62366-1 specifies that the documentation must include the planning of a formative evaluation. You won’t find such granular requirements in the MDD, MDR, or ISO 13485.
The following mind map provides a summary and overview of the documents that medical device manufacturers must prepare for a conformity assessment to demonstrate that their medical device meets the essential requirements according to Annex I of the Medical Device Directive.
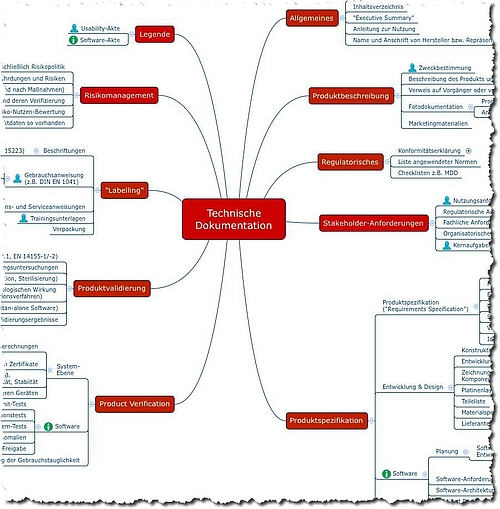
Fig. 2: Mindmap with contents of technical documentation. You can download a translated version free of charge as part of the starter kit.
3. Structure of technical documentation
Regulatory requirements do not specify how manufacturers should structure technical documentation. One exception is the FDA, which for example provides the chapter structure including chapter numbering for premarket notifications or 510(k).
a) Objectives of a “standardized” structure
Some "associations" have drawn up proposals for the structure of technical documentation to try to achieve the following objectives:
- Manufacturers can get an overview as quickly as possible as to what documentation is required.
- The authorities and notified bodies can quickly navigate and review the technical documentation thanks to the uniform and logical structure.
- Manufacturers have to spend less time re-writing the files for different legal fields.
b) Proposals for the structure of technical documentation
STED: Summary Technical Documentation
One of the best known proposals for structuring technical documentation comes from the IMDRF (formerly the GHTF). A lot of authorities and notified bodies use the STED (Summary Technical Documentation) as a guide.
ASEAN CSDT
The Association of Southeast Asian Nations (ASEAN) has also published a proposal for the structure of the technical documentation, known as the Common Submission Dossier Template (CSDT).
This ASEAN CSDT is called “Guidance on Preparation of a Product Registration Submission for General Medical Devices using the ASEAN Common Submission Dossier Template”.
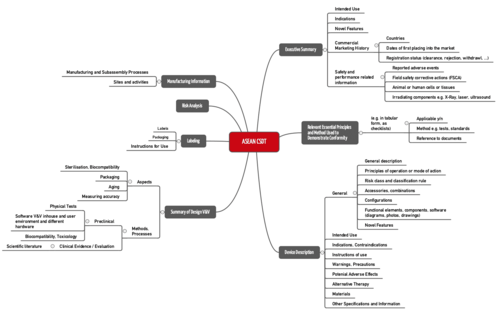
Fig. 3: Structure of technical documentation according to the ASEAN CSDT (click to enlarge)
NB-MED
The proposal of the NB team, set out in document NB-MED 2.5/1, is also relevant in practice.
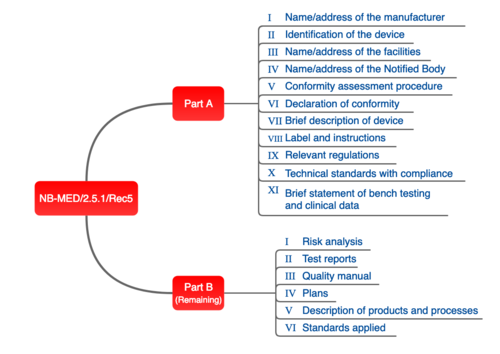
Fig. 4: Structure of technical documentation proposed by the NB team.
Johner Institute
The structure shown in Fig. 2 is particularly well suited to manufacturers of active medical devices: It is compact, easy to understand and ideal for meeting regulatory requirements.
4.Technical File for different legal systems
Many, but not all, international authorities accept standardized formats, such as STED. Nevertheless, there are some country-specific differences.
To ensure that manufacturers do not lose track of these differences, we recommend using a “mapping table,” which contains all of the required documents and everything needed to show that the essential requirements of products bearing the CE mark have been met, that need to be addressed in the respective legal systems.
Document Type | FDA submission | FDA DMR | FDA DHF | CE documentation |
Table of contents | VOL01_TOC | -- | -- | Cover sheet |
SRS | VOL 023 Appendix-B Requirements Specification | 02 Device Description | ... | ... |
... |
|
|
|
|
Fig. 5: Table comparing the documents and requirements of different legal systems
The columns, which represent the structures named above, describe where specific information can be found in each file.
Maintaining a mapping table and compiling the specific approval documents for each market is usually the responsibility of the regulatory affairs department.
5. Interaction between technical documentation & quality management systems
Manufacturers must prepare the technical documentation for their medical device and submit it to the authorities (except for class I devices). The technical documentation is also subject to FDA inspection or ISO 13485 audits. Based on this documentation, the inspector/auditor will assess whether the basic requirements of the regulations have been complied met and whether the manufacturer conforms to their quality management system.
Finally, the quality system must define the processes (e.g., development and risk management) used to develop and produce the medical device.
Therefore, the technical documentation (technical file) is not just something that manufacturers have to submit to the authorities; it is more a set of documents compiled organically throughout the development process.
In brief, without consistent, standard-compliant, and complete technical documentation, medical device manufacturers cannot prove that their medical device meets the essential requirements for approval or that their quality management system is effective. For most manufacturers, designing a functioning quality system is a prerequisite for the conformity assessment.
If you have any device/country specific questions on the topic of Technical Documentation for medical devices, please do not hesitate to drop the Johner Institute a message, we would be delighted to help.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.