Harmonized standards: Evidence for medical device manufacturers
Most medical device manufacturers benefit from harmonized standards to demonstrate compliance of their medical devices with the general safety and performance requirements.
1. Standards and harmonized standards
a) Definitions and more
EU Regulation 1025/2012 defines the term harmonized standard as "a European standard adopted on the basis of a request made by the Commission for the application of Union harmonisation legislation."
Standards are documents written by national or international standardization commissions to document the general accepted state of the art. They usually do not describe best practices or even the scientific state of the art, but rather the minimum consensus requirements that the standards committee could agree upon.
The state of the art describes the developed stage of technical capabilities at a given time. In contrast, the scientific state of the art describes the current knowledge about studied matter .
Specific encryption methods with a defined key length, for example, correspond to the state of the art. The scientific state of the art, on the other hand, knows quantum-physical procedures for encryption and decryption. They, therefore represent a level that a professional company should not fall below.
Additional information
Read more about the distinction between state of the scientific state of the art here

Some standards, the so-called harmonized standards, have been selected by the EU and published in the Official Journal mentioned above. If manufacturers comply with these harmonized standards, auditors, for example, assume that the statutory requirements are met. Specifically, the general requirements of safety and performance, see Annex I of MDR and IVDR, are met.
b) Chain of evidence
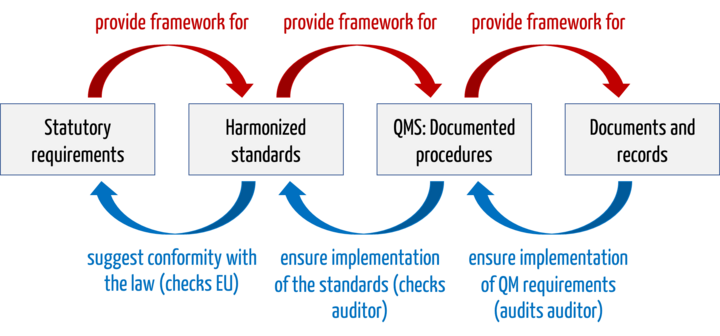
An example of this chain of evidence:
- Legal Requirements: MDR basic requirement on software lifecycle processes.
- Harmonized standard: IEC 62304
- QMS and documented procedures: Software development standard operating procedure or the software development plan, which require code review, among other things
- Documents and records: Documented code review on day X, performed by person Y with output Z.
The harmonized standards are not legally mandatory, both in the EU and with the FDA. However, without the harmonized standards, medical device manufacturers will struggle to demonstrate that their devices meet the legal requirements.
c) Examples of harmonized standards
These harmonized standards include:
- EN IEC 62304: Software life-cycle processes for medical devices.
- EN IEC 62366-1: Application of usability to medical devices
- EN ISO 14971: Application of risk management to medical devices
- EN IEC 60601-1: Medical electrical equipment and systems: Basic safety and essential performance
- EN ISO 13485: Medical devices - Quality management systems - Requirements for regulatory purposes
2. Standards in the context of MDR and IVDR: The theory
The MDR continues to recognize the concept of harmonized standards that may serve as " presumption of conformity." It writes in Article 8:
„Devices which are in conformity with the relevant harmonised standards, or the relevant parts of those standards, the references of which have been published in the Official Journal of the European Union, shall be presumed to be in conformity with the requirements of this Regulation covered by those standards or parts thereof.“
In addition to the harmonized standards, however, there are supposed to be common specifications. Article 9 of the MDR states:
„where no harmonised standards exist or where relevant harmonised standards are not sufficient, or where there is a need to address public health concerns, the Commission […] may […] adopt common specifications (CS) in respect of the general safety and performance requirements set out in Annex I […]“
This means that manufacturers will have to use multiple evidence tools in the future.
The requirements of the IVDR are analogous.
3. Standards in the context of MDR and IVDR: The practice
a) The problems with the standards
So many problems now appear that the question of their future relevance arises. Examples of the problems are:
Problem 1: Heterogeneous quality
For example, the published AI standard contains many good aspects. However, it is an example that many standards lack precise models. This makes the requirements arbitrary. Moreover, these requirements are often not sufficiently coordinated between some standards.
Problem 2: Lack of up-to-date information
The quality of the standards also depends on how up-to-date they are. The main parts of IEC 62304, for example, date back to 2005, two years before the first iPhone was released. At that time, smartphones, cloud computing, and AI were not nearly as important as today. The NSA scandal was still eight years in the future. Is a standard like this supposed to reflect the state of the art?
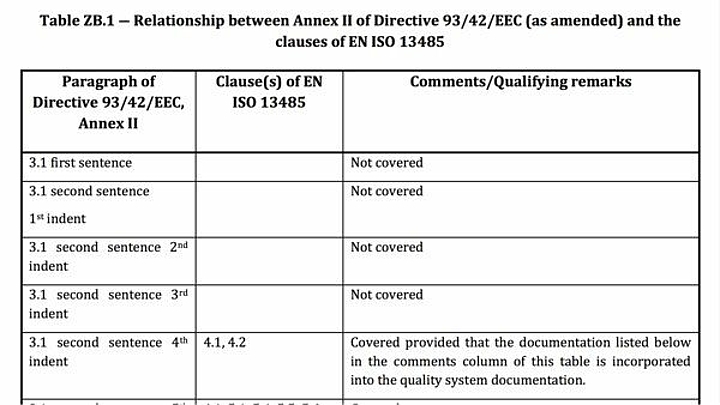
Problem 3: Lack of coverage of regulatory requirements
Even current harmonized standards usually do not address all the requirements of the EU regulations.
The "Z-annexes" provide an overview of which regulatory requirements manufacturers can assume to be fulfilled when complying with the respective harmonized standard and which they cannot.
Problem 4: Prices are too high
Standards sometimes cost several hundred euros. We monitor over 4000 regulatory documents for our customers. A notable portion are standards. Even if the typical manufacturer only needs a subset, the financial burden is still appreciable.
Tip
Estonian standards (also in English and with harmonized content) cost only a fraction of the cost.
Problem 5: Hesitant / incomplete harmonization
In 2017, the MDR came into force. More than six years later, only a fraction of the standards that were harmonized under the MDD and AIMD have been harmonized. The situation is analogous for the IVDR.
b) The causes
There are many causes that contribute to these problems:
- Bureaucracy
There are standards bodies that shut themselves down with bureaucracy. People are more busy with regulations, internal rules, international coordination and sometimes disputes than with the actual standards work. - Lack of resources/competence
This is another reason why it is not easy to attract the best in a field to work on standards. The highest level of competence is required: domain expertise, scientific methodology, the capability to develop abstract models and at the same time ensure practical relevance and feasibility in practice. - Competing preferences
The EU Commission does not give the impression that it gives the same importance to standards as it did in the past. The EU spends millions on HAS consultants, who give the impression of rather dragging out the procedure. Moreover, with the Common Specifications, the EU has created an alternative to the standards, which it holds in its own hands.
c) The consequences for manufacturers and notified bodies
The problems with standards mean that authorities and notified bodies sometimes refer to legislation, sometimes to the latest versions of standards, sometimes to harmonized versions of standards, and sometimes to other best practices and guidelines.
For manufacturers, this means:
- Legal uncertainty in audits and approvals
- More effort for subsequent improvements
- Higher costs
- And thus a longer time to market
All this happens in a competitive environment that challenges manufacturers beyond their limits.
d) The approaches to solutions
In Europe, we are therefore well advised to solve these problems. There are several approaches to this and which we should pursue all of them(!).
Returning to the original idea
It would be very helpful to speed up again, to improve the standards, to update them, to harmonize them and to offer them at affordable prices. We members of the standards committees are already working for free.
However, the likelihood of such a new start is limited as long as the will is missing and a correctly lived bureaucracy seems to be more important than the contribution to patient welfare.
Important
Nevertheless, or precisely for this reason, our request: Get involved in the standards committees!
Optimize efficiency on the manufacturer side
The tasks of manufacturers consist of more than "just" proving the conformity of their devices and processes. There are opportunities for optimization:
- The wheel does not have to be reinvented every time: When it comes to software, not only can components and frameworks be reused, but also entire backend services.
- In clinical evaluation, literature searches can be substantially accelerated. One of our customers was able to reduce the number of literature references to be evaluated from 1000 non-specific to 60 specific ones with the help of the Medical Writer course and save effort accordingly.
- Many tasks can be avoided completely through automation. More about this below.
Create de facto standards
The decision on the conformity of devices and processes is a binary one. Compliant or non-compliant. This decision arises from many micro-decisions. For example, is the required list of SOUPs available? Yes or no.
These decisions need to be mapped into algorithms - which we do. If there is an understanding on these algorithms, a de facto standard, thus legal certainty and finally the basis for automation is created.
4. Certification?
Manufacturers wonder if they need to certify themselves or their devices to a standard. The short answer is:
If a conformity assessment procedure (e.g., under Annex II of the MDR or Annex IX of the MDR) requires a quality management system, manufacturers must have that QM system certified by a notified body. If successful, they receive a certificate confirming conformity with the relevant Annex (and usually also with ISO 13485).
When reviewing the QM system, the notified bodies also check,
- whether the manufacturers work in conformity with the harmonized standards such as ISO 14971 or IEC 62304 and/or
- whether the specifications of the QM system require work that conforms to these standards.
A certificate confirming conformity to the requirements of ISO 14971, IEC 62304, IEC 62366-1, etc. is neither required nor common.
Exceptions include standards such as IEC 60601-1, but again, the manufacturer does not need to be certified. Instead, it is advisable to select a test laboratory that performs the inspections in accordance with this standard or standard family.
5. Which standard to use?
a) DIN or EN or ISO/IEC?
A frequently asked question is: Which variants of the standards should be used? The DIN, the EN or the ISO or the IEC?
Harmonized standards can be used to prove that devices or systems meet the requirements of European directives or regulations. Therefore, the European standards should be used, i.e. the EN standards. The national standards, i.e. the standards with a prefix such as "DIN EN", are identical in content and can therefore also be used.
In contrast to the "EU variants", the international standards (ISO, IEC) do not have the Z annexes. The Z-annexes contain the "mapping" of the regulatory requirements to the normative requirements (EU directives, EU regulations). This means: the Z-annexes describe which regulatory requirements are fully, partially or not at all covered by the standard.
Caution
Harmonization has nothing to do with numbering. For example, DIN EN 14971 is not the standard on risk management for medical devices (which is DIN EN ISO 14971), but one on "Textiles - Knitted fabrics - Determination of the number of meshes per unit length and unit area".
b) Which version of the standard?
There is always discussion about the transition periods. Unfortunately, there are no clear rules. But the following rules of thumb can help:
- If a standard is harmonized, then use that version.
- If the standard itself specifies a transition period, then follow it.
- Otherwise, assume a transition period of three years.
- For new devices, work with the latest versions.
- For devices already on the market, do a gap analysis no later than three years after a new version is published.
6. Conclusion and summary
Harmonized standards should help manufacturers and notified bodies gain a common understanding of how the requirements of the MDR and IVDR should be met.
Unfortunately, harmonization of standards is faltering, so this objective is only partially met. Nevertheless, manufacturers should benefit from relevant standards. After all, they describe the state of the art and help to demonstrate that regulatory requirements are met.
Change history
- 2023-09-06: Chapter 2 disassembled. Chapter 3 added.
- 2023-04-03: Article completely revised and updated. Removed the "News" and "Issues" sections and integrated them into the text where necessary.
- 2022-06-09: Added section "June 2022" in the "News" section.
- 2022-03-16: In the "News" section, added the section "January 2022".
- 2021-07-27: In the "News" section, added the section "July 2021".
- 2021-04-26: In the "News" section, added the "April 2021" section.
- 2020-11-08: Added new draft for a Standardization Request. Text adapted in this respect.

Author:
Mario Klessascheck

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.