FDA: The 510(k) clearance (Premarket Notification)
510(k) clearance is the primary procedure for obtaining marketing clearance in the United States. It is also referred to as Premarket Notification (PMN).
This article provides a quick overview of the procedure and the documents that must be submitted.
Further information
Note an overview of articles on the FDA and its approval process.
1. General information on the 510(k) clearance by the FDA
In contrast to the European jurisdiction with the conformity assessment procedures, the FDA requires an explicit approval or clearance of medical devices. The most prominent procedure is named after an article of the Food, Drug, and Cosmetic Act (FD&C):510(k).
Manufacturers can generally use this procedure when their devices fall into Class II, and there is a comparable device that has already been cleared. This device, known as the predicate device, must also actually be comparable. This is referred to as substantially equivalent.
Learn more about "predicate devices" below.
The 510(k) procedure is also applicable to certain class I devices that are not "510(k)-exempt," as well as a small number of class III devices.
The 510(k) variants
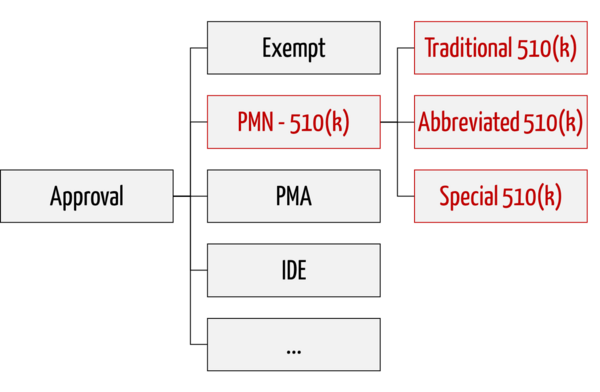
The FDA distinguishes between three variants of premarket notifications:
1. Traditional 510(k)
The traditional procedure is used for "me too" devices and serves as a fallback if the FDA does not recognize Special 510(k) and Abbreviated 510(k).
2. Special 510(k)
Special 510(k) is an expedited procedure that can be used when changes are made to a user's own device.
3. Abbreviated 510(k)
Abbreviated 510(k) is a procedure that relies on compliance and adherence to standards, "Special Controls," and "Guidance Documents." A special form of this procedure is the novel Safety and Performance Based Pathway.
Further information
Read more about Abbreviated 510(k) here.
2. Content and format of the submission documents
To increase the efficiency of the 510(k) procedure, the FDA has now introduced an electronic submission format known as the eSTAR format. The FDA only accepts 510(k) submissions via eSTAR as of Oct. 1, 2023. This is an interactive submission in PDF format. It covers the requirements of 21 CFR part 807 Subpart E and is based on the IMDRF requirements, more specifically, the "Table of Contents" format. For each section in the template, the number of the corresponding chapter of the Table of Contents structure is given. This is very positive and brings us a small step closer to harmonizing a worldwide submission format. It is a pity that the EU Commission did not also follow this approach for Annex II of the MDR on technical documentation.
The eSTAR template to the 510(k) (non-IVD) follows this structure:
- General Introduction (e.g., version history of the template)
- Application/Submission Type (e.g., traditional, abbreviated, special)
- Administrative Information (contact details, reference to pre-submissions, list of applied standards)
- Device Description (detailed product description)
- Indications for Use (statement on the intended use/indications of the device)
- Classification (information on the classification of the device, including regulation number and product code)
- Marketing History (information on the market history)
- Predicates and Substantial Equivalence (detailed comparison with the selected predicate device(s))
- Labeling (labeling and instructions for Use)
- Reprocessing, Sterility, and Shelf-Life (information on reprocessing, sterilization, and shelf-life, as applicable)
- Biocompatibility (information on biocompatibility assessment, if applicable)
- Software/Firmware and Cybersecurity/Interoperability (software documentation including cybersecurity and interoperability information, as applicable)
- EMC, Wireless, Electrical, Mechanical, and Thermal Safety (evidence such as test reports on the above aspects, as applicable)
- Performance Testing (evidence of further tests, such as bench, animal or clinical tests)
- References (e.g., to referenced technical literature)
- Administrative Documentation (e.g., Truthful and Accurate Statement, Declaration of Conformity, 510(k) Summary or Statement)
Further information
For guidance on what to include in each chapter, see the Format for Traditional and Abbreviated 510(k)s guidance.
3. Predicate Device
A predicate device is a medical device already (legally) marketed in the U.S. that medical device manufacturers can refer to for clearance. Such a predicate device is a prerequisite for the 510(k) clearance preferred by manufacturers, which is less complex than a PMA (premarket approval).
Proof of equivalence
In order for the manufacturer to be allowed to cite this predicate device, he must prove that it is substantially equivalent.
In most cases, however, manufacturers do not want to market an identical device (e.g., the umpteenth syringe) but one that has the same intended purpose but differs in technical implementation.
The revised guidance Evaluating Substantial Equivalence in Premarket Notifications gives you very specific guidance on when you may cite a device as "substantially equivalent" and thus reference it as a predicate device for a Premarket Notification. In doing so, the FDA looks at the following:
- Do the devices have the same intended purpose?
- Do no new safety or effectiveness issues arise?
- Are the devices equivalent (technically, biologically, etc.)?
4. Criticism
The approval procedure is based on the existence of a predicate device. The risk-benefit ratio has already been evaluated for this device. A re-evaluation, therefore, usually no longer takes place.
However, this predicate device can itself have a predicate device. In this way, entire chains of references are created, which ultimately end with medical devices that first came onto the US market decades ago. This means that the current state of the art is no longer guaranteed.
This was already criticized by John Oliver many years ago (see video).
The FDA is aware of this problem and is working on new approval procedures.
In September 2023, the FDA initiated initial steps to modernize the 510(k) process and published a series of guidance documents. You should pay particular attention to the Best Practices for Selecting a Predicate Device to Support a Premarket Notification [510(k)] Submission. With these "Best Practices," the FDA imposes stricter requirements on manufacturers when selecting a Predicate Device. Accordingly, selecting any equivalent device as a predicate is no longer possible. Manufacturers must now conduct more detailed research and only choose comparative products that have been developed and manufactured according to current (recognized) standards and ideally have no design-related recall history.
5. Summary and conclusion
The 510(k) is still the most popular approval process in the USA. It is clearly described and predictable, and when all documents are available, the FDA is obliged to make a decision within 90 days.
However, this leads to manufacturers approving "me too" devices rather than innovative products. Therefore, the approval process will become less important in the future.
Further information
Also see our article Avoiding the Five Most Common Mistakes When Submitting a Premarket Notification.
Support
The regulatory affairs experts at the Johner Institute provide support for the worldwide approval of medical devices. Feel free to get in touch, for example, via the contact form.
The Medical Device University explains step by step
- the requirements of the FDA,
- how Predicate Devices are searched for and justified,
- how to prepare the submission file and
- what manufacturers have to do during "registration," "listing," and after placing on the market.
Change History
- 2023-10-24: Reference to a new article on "Predicate Device" and "Substantial Equivalence" added.
- 2023-09-14: Chapter 2 revised (eSTAR format). Latest information added to Chapter 4 on modernization of 510(k) procedure.

Author:
Luca Salvatore

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.