Abbreviated 510(k) - When the abbreviation is allowed
Abbreviated 510 (k) is one of three 510(k) clearance processes offered by the FDA.
This article tells you,
- what work you can save on admission by "abbreviation."
- which requirements you have to fulfill for this and
- which is why the term "abbreviated" can be misleading.
1. Abbreviated 510(k) and other approval procedures
If manufacturers want to market their medical devices in the U.S., they must go through one of the approval processes. Among the most commonly used are the Premarket Notifications PMN. These are better known as 510(k) about the corresponding article in the Food Drug and Cosmetic Act. In these 510(k) processes, FDA distinguishes three:
- Traditional 510(k): The classic approval process for "me too" products
- Abbreviated 510(k): The abbreviated approval process under certain circumstances.
- Special 510(k): Simplified re-registration of own product(!) in case of product changes
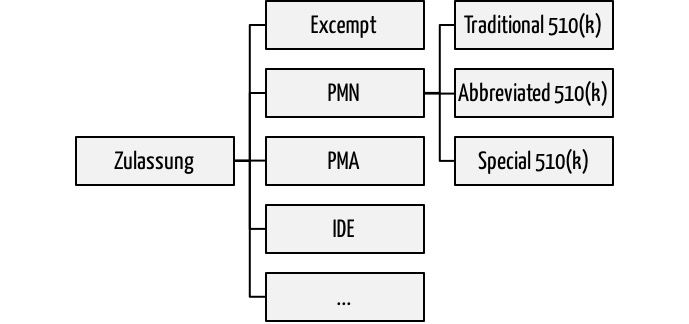
Fig. 2: The proof that a product is as safe and efficient as an equivalent product should increasingly also be possible indirectly, e.g., via standards. This is the aim of the "Extended Abbreviated 510(k)".
Further information
Note the Guidance Document der FDA "The Abbreviated 510(k) Program ".
2. Abbreviated 510(k): What you save compared to Traditional 510(k).
No shortened duration
The term "abbreviated" suggests that the approval process would be shorter. Unfortunately, this is not entirely true. A shortened duration of 30 days applies to "Special 510(k)s" but not to "Abbreviated 510(k)s", and the latter has the same time as the "Traditional 510(k)".
Less documentation
The hope that the documentation for an "Abbreviated 510(k)" would be shorter than for a "Traditional 510(k)" also seems to be deceptive at first glance. The format of both approval procedures, i.e., the chapter, structure, does not differ(!).
Instead of detailed documentation, manufacturers may submit summaries referencing standards complied with (declarations of conformity) for an Abbreviated 510(k). I.e., the chapter "9 Declarations of Conformity and Summary Reports" can be somewhat more extensive for an Abbreviated 510(k), chapters 14 to 19 can be shorter.
Because this means less documentation for an FDA reviewer to review, a decision can be made more quickly than a Traditional 510(k). The only thing is that the agency does not want to commit to this.
Equal costs
Those hoping that lower agency overhead will translate into lower fees will be disappointed. The costs are the same for all 510(k) procedures.
3. When you may use an Abbreviated 510(k)
Requirements
An Abbreviated 510(k) is allowed when one of the following three conditions is met:
- The FDA has published a guidance document for the relevant product or technology.
- The manufacturer applies the "Special Controls." Examples are post-market surveillance compliance with development guidelines or specific standards. Which of these Special Controls are to be followed is governed by the "Product Classification Database."
- The FDA has recognized relevant standards that the manufacturer follows, and the FDA maintains over 400 such "recognized standards" in its database.
Examples
- For example, the FDA lists 15 "Consensus Standards" for nuclear devices. A quality management system is mandatory, and further special controls are not explicitly mentioned. However, an overview page lists further standards and guidance documents.
- For implantable RFID transponders, the FDA calls them "Special Controls", for which they have even published their guidance document.
- For digital mammography systems, the FDA lists four standards and refers to the associated guidance document with the special controls. These controls include electrical safety testing (according to IEC 60601-1), biocompatibility testing, phantom testing, and many more.
A European approach
The idea behind Abbreviated 510(k) is that FDA does not have to use the manufacturer's documentation to assess whether the product meets regulatory requirements, such as product safety. Rather, the agency assumes that this is the case if the manufacturer meets or follows the relevant standards, guidance documents or other best practices.
This is in line with the European Medical Device Directives and Regulations approach, allowing a presumption of conformity if manufacturers follow harmonized standards and common specifications (Common Specifications).
4. Course of the procedure
Declaration of Conformity
If the manufacturer applies a "recognized standard," i.e., declares conformity with a standard (or several standards), he must do so in the form of a "Declaration of Conformity" containing the following information:
- Standard(s) (clearly identified)
- The declaration that the standard(s) have been complied with (except for non-applicable requirements).
- List of non-applicable requirements
- Explanation of all deviations when applying the standard, e.g., when testing
- Description of any differences between the tested device and the devices that will be placed on the market
- Contact details of the test laboratory
The FDA emphasizes that the manufacturer bears compliance and cannot shift it to a testing laboratory.
Rejection
If FDA deems a manufacturer's 510(k) premarket notification ineligible for the "Abbreviated" program, the agency offers to convert the process to a "Traditional" process. However, the manufacturer must then assume that FDA will request additional documents.
5. Draft Guidance Document "Expansion of the Abbreviated 510(k) 2 Program."
Objective
On April 12, FDA released a draft of a new guidance document. It is titled "Expansion of the Abbreviated 510(k) Program: Demonstrating Substantial Equivalence through Performance Criteria." The goal is to reduce the 510(k) procedures burden on both manufacturers and the agency.
This effort arises primarily from the need to prove that the new product to be approved is as safe and effective as a comparator product, the "predicate device." Increasingly, the FDA wants to make it possible for manufacturers to prove equivalence, not by direct comparison and direct measurement. Instead, they should be allowed to refer to standards, "special controls," and guidance documents.
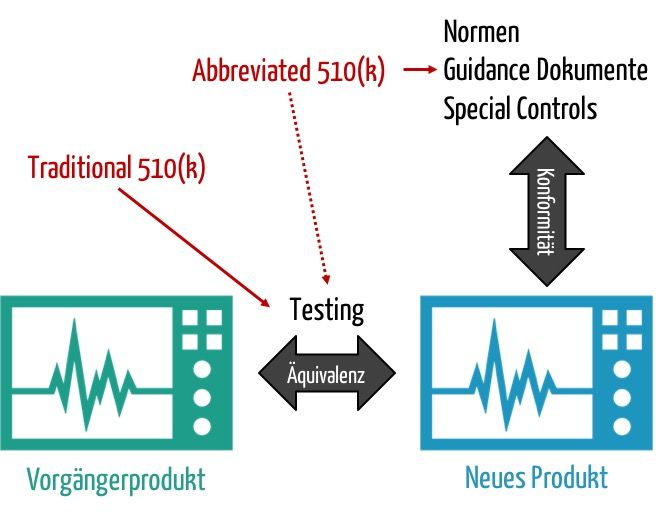
Fig. 2: Proof that a product is as safe and efficient as an equivalent product should increasingly also be possible indirectly, e.g., via standards.
The FDA thus intends that even more approvals will be possible via the "Abbreviated Program." However, the agency does not plan to change the format of the documents.
Requirements
As a condition for manufacturers to participate in this expanded "abbreviated" program:
- The new product has an intended purpose and technical characteristics that do not raise new safety and efficacy issues beyond the equivalent product.
- The performance criteria are the same as those of the legally marketed product(s) of the same type.
- The product also meets these performance criteria.
Products covered by this extended approval procedure
To determine whether a medical device may be approved under this expanded program, FDA offers assistance at the Pre-Submission Meetings.
In the future, the FDA would like to publish a list of products that are suitable for this expanded program. It would also like to specify the associated requirements (standards, special controls, guidance documents) for each product.
6. Conclusion and summary
Especially manufacturers who follow standards should strive for an "Abbreviated 510(k)" procedure. This saves them the effort of recompiling many documents for the FDA and providing evidence that has already been provided, for example, by an inspection by a test house.
A prerequisite is that the FDA lists the standards as a "recognized standard." Alternatively, or in addition, manufacturers must observe the FDA's guidance documents.
The procedure is particularly suitable for European manufacturers working according to harmonized standards. Even if the FDA does not recognize the "Abbreviated 510(k)" route, nothing is lost, and the procedure can be continued as "Traditional 510(k)".
The new guidance document on Abbreviated 510(k) indicates that the FDA intends to increase the importance of this approval procedure. This is very welcome from the manufacturers' point of view.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.