PMCF studies: three types to be distinguished
PMCF studies are studies that manufacturers use as part of post-market clinical follow-up to continuously demonstrate compliance of their medical devices.
Manufacturers do not always need to conduct studies to meet PMCF requirements. And not all types of PMCF studies are subject to MDR requirements.
This article compactly summarizes the regulatory requirements in the context of PMCF studies and provides tips on how to gain legal certainty and avoid unnecessary efforts and detours in PMCF.
1. PMCF studies: the basics
a) Definition
PMCF studies are clinical investigations within the meaning of Article 74 MDR that are conducted after the product has been placed on the market. At this point, the product already bears a CE marking.
A clinical investigation is defined by the MDR as a "systematic investigation involving one or more human subjects conducted for the purpose of evaluating the safety or performance of a device."
The type of clinical investigation determines the regulatory requirements. These are presented in the second chapter.
b) Objectives of PMCF studies
Demonstrate safety and performance of medical devices
The objective of the post-marketing clinical follow-up of a product is to collect additional clinical data on one's product after it has entered the market in order to continuously evaluate the safety, performance, and efficacy of the product.
PMCF studies are intended to answer questions such as:
- Are the assumptions on the lifetime of the medical device still correct?
- Are there (new) risks that have not yet been identified and/or accepted in the risk management file?
- Is there an off-label use of the product?
- Does the product actually achieve the expected benefit?
- Is the risk-benefit ratio still acceptable?
Meet regulatory requirements
Laws such as the MDR oblige manufacturers to proactively conduct clinical follow-up. Therefore, legal obligation is a further motivation for manufacturers.
The second chapter describes in detail which requirements the manufacturers have to fulfill.
Note
The requirements for PMCF studies and how to handle them are similar to in vitro diagnostics. For more information, please refer to our article on PMS.
2. Regulatory requirements for PMCF studies
a) At EU level
Article 61 (11) of the MDR requires that the clinical evaluation and associated documentation be updated throughout the entire life cycle of the medical device based on clinical data. This implementation should be documented in the manufacturer's post-market clinical follow-up plan (PMCF plan) according to Annex XIV Part B and the post-market surveillance plan (PMS plan) according to Article 84.
Depending on the study design and intention of the study, different regulations do apply. There are essentially three basic study types for PMCF (Fig. 1).
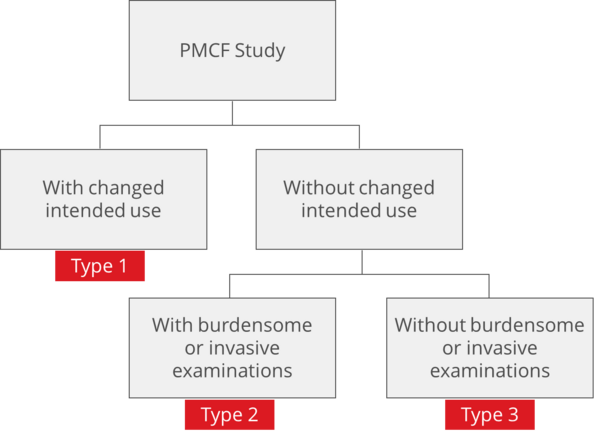
Type 1: Application of the product outside the intended use
Medical devices intended to be used in the clinical investigation outside the intended use deposited in the CE declaration of conformity are regulated according to Article 74 (2).
Example
This would be the case for an extension of the indication of a medical device or an extension of the patient population.
Thus, according to Article 74 (2), the requirements of Articles 62-81 MDR and Annex XV apply in full (requirements of an Article 62 pivotal study to demonstrate safety, performance and clinical benefit).
If the study takes place in Germany, those responsible must also take into account the MPDG §§ 24 to 30 and 62 to 70. For such studies, notification is made via DMIDS (formerly DIMDI) to the locally responsible ethics committee. These are reviewed by the higher federal authority (BfArM). The BfArM has deposited instructions for sponsors on how clinical investigations are to be recorded in the DIMDS.
Otherwise, the national laws of the country where the study centers are located apply.
Type 2: Within the intended use but with burdensome or invasive investigations
Medical devices that are intended to be used within the intended use as part of the clinical investigation but are planned to undergo additional stressful or invasive investigations due to the study design are regulated under Article 74(1) MDR. This includes procedures that go beyond those performed under normal conditions of use of the device and are study related.
Example
The MDCG 2021-6 guideline provides examples of procedures classified as "burdensome" or "invasive" at question 9. A blood draw or examination through a body orifice is considered invasive.
MDCG writes further:
„Additional procedures which are burdensome can include a wide variety of different interventions, this may include procedures which may cause pain, discomfort, fear, potential risks or complications/side-effects, disturbances of lives and personal activities, or otherwise unpleasant experiences. It is mostly determined from the perspective of the person bearing the burden.”
These studies must implicitly fulfill the requirements of Article 62 MDR Paragraph 4 b to k, m, 75 to 77, 80(5) and Annex XI, as well as contents of the MPDG. According to the German MPDG § 85 paragraph 2 No. 6, this is also a notification procedure to the higher federal authority requiring approval, which is communicated by the sponsor via the electronic system mentioned in Article 73 (DMIDS), since the procedure via EUDAMED mentioned in Article 78 of the MDR is not yet available.
A receipt of the notification of the clinical investigation shall be made by the higher federal authority. The notification has to be made by the sponsor at least 30 days before the start of the clinical investigation.
Further information
More detailed information from the BfArM on the legal requirements and further details for PMCF studies can be found on the BfArM website.
Type 3: Within the intended use and without onerous examinations
Most PMCF studies take place within the intended use and without additional burdensome and invasive investigations. The MDR does not apply in these studies, but the national rules and regulations apply.
This is an optional professional consultation in Germany according to § 15 “Berufsordnung der Ärzte”. The study must be reported to the local ethics committee.
Further information
Note the article on PMCF and the need to involve the ethics committee.
In Germany, these studies are also called BO-Ä studies. You can find out what you need to consider and which application documents you need in the guide "Regulatory classification of clinical investigations with medical devices" (only available in German) from the Federal Ministry of Research and Education.
Summary
Medical Device | Without CE | With CE (after placing on the market) | ||
Use | Before placing on the market | Outside the intended use | Within the intended use | |
With additional and burdensome examinations | Without additional and burdensome examinations | |||
Type |
| Type 1 | Type 2 | Type 3 |
Specific requirements of the MDR | MDR Art. 62 | MDR Art. 74 (2) | MDR Art. 74 (1) | — |
Further regulatory requirements | MDR Art. 62-80 + Annex XV In Germany also MPDG §§ 24–30, 62–70 | MDR Art. 62 Par. 4b–k, m, 75–77, 80 (5) und Annex XV + MPDG (GER) | No application of MDR or MPDG; national regulations apply if necessary | |
Standards/ Guidelines | DIN EN ISO 14155 - 2021-05 Good clinical practice for clinical investigations of medical devices Declaration of Helsinki, current version from October 2013, Fortaleza (Brazil) | |||
MDCG clinical investigation guidelines: 2023/C 163/06, MDCG 2021-28,MDCG 2021-20, MDCG 2021-8, MDCG 2021-6 MDCG 2020-10/1 MDCG 2020-10/2. | IMDRF Guidance PMCF Studies: IMDRF MDCE WG/N65FINAL:2021 | |||
Objective | Conformity assessment | Ongoing evaluation of the safety, performance and clinical utility of the product | ||
Tab. 1: Overview of regulatory requirements for PMCF studies
b) At national level
Depending on the country in which the clinical investigation is conducted, country-specific requirements apply.
Germany
The EU regulations (MDR and IVDR) have also brought about a renewal of national regulations and laws. These adaptations, as well as specific national provisions, were mainly made by the Medical Devices EU Adaptation Act (MPEUAnpG). Under Article 1, the MPEUAnpG contains the Medical Devices Implementation Act (MPDG), which replaces the previously valid Medical Devices Act as of May 26, 2021, for medical devices covered by the MDR.
Necessary changes for in vitro diagnostic devices are already included in the MPEUAnpG; however, these will not enter into force until May 26, 2022, in a deviating manner. For more information, see our blog article on the Medical Device Law Implementation Act (MPDG).
Austria
The Austrian Federal Law for the Regulation of Medical Devices (Medical Devices Act 2021 - MPG 2021) has been published in a new version. The information can be found in the Federal Legal Information System (RIS), a system that provides the laws in Austria online.
The consolidated Austrian MPG is very similar to the MPDG. The important passages on clinical testing are deposited in Section 3 §§ 13-36. Other important laws can be found in the BASG, the Medical Device Operator Ordinance and the Medical Device Notification Ordinance. Similar to Germany, there is also an Austrian Medical Device Operator Ordinance.
Switzerland
Switzerland has adapted the regulation for medical devices to the EU rules. This was done in the interest of patient safety and EU market access for the Swiss medical device industry. The Ordinance on Clinical Investigations with Medical Devices (KlinV-Mep), published on the website of the Swiss FOPH, clearly lists all regulations on research with medical devices in one legal text.
The entry into force of the adapted human research regulations has been set for May 26, 2021. The websites of Swissmedic and swissethics provide up-to-date information on applications for clinical investigations involving medical devices.
France
The national guideline for medical devices in France is set by the Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM). The implementation of the MDR is regulated in France in the regulation 2022-582 from April 2022. Important when submitting: Labeling, declaration of conformity, and instructions for the use of medical devices must be in French, according to the French Public Health Code.
Spain
The Directorate General of Medicines and Medical Devices (Dirección General de Farmacia y Productos Sanitarios) is part of the Spanish Ministry of Health (Ministerio de Sanidad).
Royal Decree 1090/2015, which regulates clinical investigations with medicinal products, the ethics committees for research with medicinal products, and the Spanish Clinical Investigation Registry, also issues guidelines here on the handling of medical devices, which are based on European case law.
Sweden
Läkemedelsverket (LV) is the Swedish authority for medical technology based in Uppsala. It is the approval and supervisory authority for medicinal products in Sweden and is responsible for monitoring cosmetics, hygiene products, and medical devices. As the approval and supervisory authority for narcotics, it regulates and monitors their handling and circulation.
Denmark
In Denmark, the Danish Medicines Agency is authoritative for the national guidelines. The Danish Regulation on Medical Devices and Products without medical use (BEK No. 957 of 29.04.2021) is the national implementing law that will apply from May 26, 2021. This regulation sets national requirements that economic operators must follow if they want to market devices in Denmark.
There is an obligation to provide labels and instructions for use in Danish when the product is made available to the end-user or patient. However, this requirement may be excluded by the Danish Medicines Agency in exceptional cases (Art. 3, para. 2 BEK), e.g., taking into account the user's professional and linguistic requirements for using the device with instructions in a foreign language (mostly English) and the intended use of the device.
In principle, the declaration of conformity can be drawn up in English. Nevertheless, the Market Surveillance Agency may require a translation into Danish (Art. 5 BEK).
3. Typical mistakes in the planning of PMCF studies
Mistake 1: Too many or the wrong study centers
Selecting a suitable study center can prove to be a difficult undertaking. Yet the right choice can have a decisive influence on data quality. A competent and high-quality center is the basis of a good study.
Good study centers can be recognized by the equipment of the study site, the adherence to specified time frames, high capacities for patient recruitment, experienced staff, references of already conducted studies, and finally also by the called costs.
Collaborating with multiple study sites can help you gather the data you need much faster. However, be aware that each additional study center will need to be trained, audited, and equipped and that you will need to fulfill your sponsorship obligations for all centers.
Mistake 2: Too many countries of study
Similar to the study sites, the choice of study countries is also decisive for the timing and costs of a clinical investigation. Find out exactly about the national regulations and conditions in your target countries and take note of any special features in the regional submission.
The goal of the study should be to use clinical data to demonstrate your outcome parameters within your established acceptance criteria. The extent to which genetic alterations or regional ethnic divergence are necessary to support your study data can be determined from the state of the art.
Mistake 3: Incorrect study design
As with any high-quality clinical study, it is important to choose the correct study design for PMCF studies in order to properly collect data. In doing so, the study design can be varied depending on the requirements (e.g., RCT studies or simulation studies, "open-label" studies or cross-over studies, intervention studies or observational studies). Which study design is fitting for you can only be addressed case-specific to your product. The right choice will save you time, money, and nerves.
Mistake 4: Misjudgement of the time required
Many manufacturers underestimate the time required for patient recruitment when planning their studies. Particularly in the case of more highly classified products or when it comes to access to the corresponding patient populations and schedules, difficulties arise. This is particularly significant in the case of rare diseases or small study populations, where the initial populations can be very small.
Mistake 5: Inadequate case number planning
In order to obtain valid results, planning and calculation of the number of cases are absolutely necessary before any data collection is carried out. In the case of clinical studies and animal experiments, this is even mandatory and is closely checked before approval is granted (MDR, Regulation EU 745/2017 Annex XV, Chapter 1, Section 2.1; DIN EN ISO 14155:2021-05). Even in other areas of application, a lack of case number planning does not speak for the quality of the study. Failures can be associated with enormous costs or long study times.
Case number planning depends on the primary research question and the planned statistical analysis method. A number of assumptions regarding parameters should be made in advance (e.g., acceptance criteria, available clinical data, definition of endpoints, and statistically manipulated variables). Each case number planning is, therefore, individually tailored to the respective study and its situation.
Mistake 6: Lack of clarity about regulatory requirements
With the abundance of requirements of the MDR and the MPDG for the different types of studies, it is difficult to keep track of everything, especially if you are planning your first clinical investigations or PMCF studies. Extensive assistance in implementing the requirement for planning and conducting clinical investigations is provided by the BfArM on its homepage and by the Association of Medical Ethics Committees.
The BfArM guideline (only available in German) helps to classify the products correctly.
Mistake 7: Incorrect selection of a Clinical Research Organization (CRO)
Clinical Research Organizations (CROs) can assist you with all of the above. However, most CROs are specialized in drug research.
Therefore, our tip!
It is not always necessary to use a (possibly expensive) CRO. After all, not every product needs a clinical investigation or PMCF study. You can learn more about this in our articles on MDCG 2020-6, clinical evaluation, or in our seminar on clinical evaluation.
4. Tips for planning PMCF studies
Tip 1: Standardize the evaluation of data
Standardized processes facilitate data evaluation enormously and create clear structures for technical documentation and auditing. Here, the data should be organized uniformly across all devices, medical indications, and target populations. This makes it easier to see which data is available and which still needs to be collected.
Additionally, it helps to ensure that all areas are covered in the division of labor. A standardized process also makes it easier to track other risk factors for manufacturers, such as complaint trends, recalls, or changes in specific marketing regions.
Tip 2: Prioritize product portfolio
As a manufacturer, analyze your medical devices according to the following criteria:
- Sales, contribution margin
- Product impact in the medical market
- Expiration dates of certificates
- Results of gap analyses
- Quantity of products to be tested in relation to the life cycle of the medical device
Derive whether the necessary PMCF activities make economic sense for all existing product classes.
Tip 3: Determine data volume and evidence level
Once all data has been collected, manufacturers should question the quality and relevance of their data from the perspective of a notified body. For devices such as implants, for example, manufacturers must include data that covers the entire life cycle of the device.
This is a good time to question the objectivity of the data collected and determine if existing biases may need to be corrected or if limitations of the data need to be outlined in the PMCF report.
Verify that sufficient clinical evidence has been collected. The key factors here are the indication, the risk class of your product, data on equivalent and similar products, and comparative data from the prior art.
Tip 4: Involve affected departments
To ensure long-term compliance, involve other departments from the beginning and keep them up to date. Business departments not involved in product regulation may not fully understand why established products incur additional or higher costs.
All departments should, therefore, be involved in discussions about product portfolios, emerging risks, and any available product data sources. As a basic principle, potential business damage should be pointed out.
Tip: Contact us now
Send an e-mail to the Johner Institute's clinical experts with your question and/or a proposed appointment for a free consultation. You will receive the necessary information to assess your project and start planning.
5. Support with PMCF studies
The Johner Institute's clinical experts will assist you in deciding whether PMCF studies are necessary at all, as well as in planning and conducting these studies.
a) Establish clinical strategy for the PMCF studies
If a study is needed, you will receive assistance in determining the clinical strategy for your PMCF study and PMCF measures in your Clinical Evaluation Plan (CEP). These include:
- Identify state of the art and derive clinically relevant parameters
- Define acceptance criteria, clinical benefit, and the methods to demonstrate it
- Assess current data and plan appropriate PMCF activities
- Plan and prepare PMCF studies
Thus, you can be confident that you have adequate study planning in place and focus on the right parameters and clinical endpoints for your PMCF study.
b) Create study design and perform case number planning
The clinical strategy will show you which type of study you need or how you can reach your goal, namely the approval of your medical product, as quickly as possible. Use the experienced experts of the Johner Institute for this. They will make sure that your costs for studies, case number planning, and submission remain as low as possible without compromising on the quality of the proof of the clinical benefit of your medical device.
This will allow you to
- know the duration and cost of clinical investigations early on and decide whether to proceed,
- be certain that your notified body or authority will accept the results at the end of the study.
6. Summary and conclusion
Legislators and manufacturers want to ensure that medical devices are safe, perform well, and are effective over the complete life cycle.
PMCF studies are an important tool to provide this evidence. However, this does not mean that PMCF studies are always mandatory. The most beneficial study is the one that does not need to be done, for example, because the data can be collected in other ways.
If a PMCF study is necessary, it is important to distinguish between the different types and to identify and fulfill the respective regulatory requirements. Neither of these is entirely trivial, but they are easily achievable with targeted help.
Tip
Send an e-mail to the clinical experts of the Johner Institute with your question and/or a proposed appointment for a free consultation. You will receive the necessary information to assess your project and start planning.

Author:
Dr. Johannes Goldmann

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.