PMS data: Do you need to involve an ethics committee when collecting data?
The MDR requires medical device manufacturers to conduct post-market surveillance (PMS) and thus collect PMS data. This article is intended to help clarify the regulatory requirements manufacturers must consider when collecting these PMS data. Notably, it is intended to highlight whether manufacturers need to consult an ethics committee in Europe if the data arise from routine product use.
1. PMS data: The question:
This article was triggered by the argument between a medical software manufacturer and a notified body. Both disagreed on the need to follow the Declaration of Helsinki and obtain ethics committee approval when collecting PMS data. There was also controversy as to whether this collecting of data already constituted a clinical trial.
In this specific case, the manufacturer used data collected during routine product use for post-market surveillance. Specifically, the manufacturer did not intervene in the treatment nor store additional data. Rather, the data came from the medical device when used for its intended purpose and were stored in its database.
The main issue to be resolved is: “When do manufacturers need ethics committee approval during PMS data collection, and when does that data collection amount to a clinical trial?”
To answer this question, it must be broken down into 8 specific subquestions. Below, you will find these subquestions and their pertinent answers.
2. Significance of PMS data
With the transition to MDR, manufacturers are becoming more aware of PMS data collection. Notified bodies require PMS plans and seek to understand how data are collected, evaluated and assessed, and how they are incorporated into, e.g., clinical evaluation.
If manufacturers (mistakenly) perceive the regulatory hurdles to collect PMS data to be too high, this may prevent them from using routine data.In doing so, they thwart theMedical Device Regulation (MDR) objective of obtaining reliable information on the safety and performance of medical devices through post-marketing data.
When the regulatory requirements are clear, manufacturers can avoid unnecessary regulatory risks (during the “approval” process). Moreover, manufacturers and notified bodies would be spared unnecessary and time-consuming conflict – and the ethics committees would not have to address needless inquiries.
3. Answers to the questions about PMS data collection
Disclaimer & acknowledgment
This article was co-authored by lawyers from a German authority and the EU who, however, do not wish to be identified. The representations in this article are a “work in progress,” as even the lawyers have not yet reached agreement on all points.
Assumptions
The answers are based on the following assumptions:
- The manufacturers collect data (or have them collected, e.g., by the operator or user). They use these data for post-market surveillance.
- These data are generated “automatically” during the intended and routine use of the product. And do so regardless of whether the data are needed for post-market surveillance. In other words, no additional data are acquired for PMS.
- No procedures are changed, no additional procedures or even procedures impacting the patients take place.
- The collected data can be assigned to individual persons (patients).
Caution
Note that all statements in this article are based on the above assumptions.
Summary
The flowchart provides a brief overview of the questions and answers:
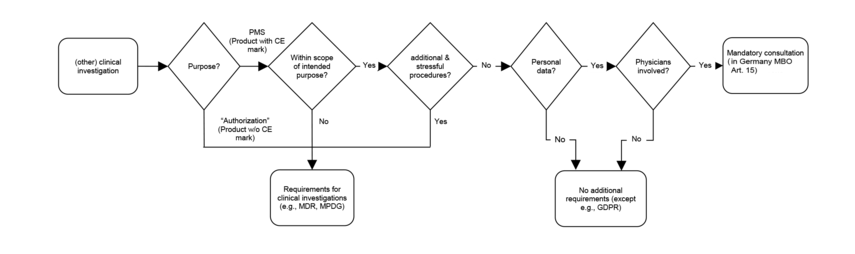
Figure 1: Decision tree for PMS data processing
When collecting PMS data under the above conditions, the MDR requirements for clinical investigations apply only to a rather limited extent. There is no need for ethics committee approval.
Data protection must always be ensured.
The German Federal Institute for Drugs and Medical Devices (BfArM) even states ( page 25) “Neither the MDR nor the German Medical Devices Law Implementation Act (MPDG) specify requirements for post-market clinical follow-up (PMCF) studies. (Meaning of the last sentence in MDR Article 74 (1): not generally applicable.)”
Figure 1: Are these data clinical data?
Answer: Yes
Reason: By definition (MDR Article 2 (48), fourth indent), these are clinical data.
Figure 2: Does this constitute a clinical investigation?
Answer: Controversial
Reason: Some lawyers argue that under MDR Art. 2 (45), this constitutes a clinical investigation because human participants are involved in the use of the device within its intended purpose, and because the objective of the PMS is to evaluate the safety and performance of the device (after it has been placed on the market).
However, there are also arguments against this view. For example, whether patients should be considered participants of a investigation is a matter of debate.
Caution!
Even if by definition PMS data collection were to be understood as a clinical investigation, it does not yet follow that (all) the MDR requirements for clinical investigations must be observed (see also question 4).
The MDR is intended to protect patients from unsafe medical devices and unethical clinical investigations. There are no such dangers when collecting PMS data.
Figure 3: Does this constitute some other clinical investigation?
Answer: No
Reason: There is no other clinical investigation as defined by the German MPDG (“sonstige klinische Prüfung”) because collecting PMS data is a systematic and planned process for product surveillance and must be carried out in accordance with the MDR. This is exactly what the German MPDG rules out in its definition of “other clinical investigations” (MPDG Article 3 (4) letter a)).
Figure 4: Do the requirements of MDR Art. 62 ff. have to be met?
Answer: Yes, but only some sections
Reason: The requirements of MDR Art. 62 only take full effect if the clinical investigation is conducted as “part of the clinical evaluation for conformity assessment purposes.”
Article 74 addresses clinical investigations of medical devices already bearing the CE mark. It requires sponsors to comply with some sections of Art. 62, even if patients are not subjected to additional and invasive or distressing procedures and the product is used within its intended purpose.
These requirements (including Article 62(4)(b)-(k) and (m)) represent general ethical and regulatory principles. However, they do not require the manufacturer to apply for approval of a clinical investigation.
Figure 5: What other regulatory requirements must be considered?
Answer: Besides the requirements of the data protection laws (General Data Protection Regulation (GDPR)and German Federal Data Protection Act (BDSG)), the professional code of conduct of the respective physicians associations in Germany must also be observed.
In addition, manufacturers must meet the requirements for
- post-market surveillance (PMS) according to Art. 83 and Annex III, and
- post-market clinical follow-up (PMCF) according to Art. 61 (11) and Annex XIV Part B.
This resulted in the issues addressed in this document.
Reason: PMS activities in which physicians participate are subject to Article 15 of the professional code of conduct ("Berufsordnung”) of their respective physician associations. These are applicable because personal data are collected. This article mandates that physicians must consult(!) with an ethics committee.
It is immaterial whether the data were already collected anyway. The key aspect here is that data collected in the course of normal use of the product are now to be used for research purposes.
If no physicians are involved, e.g., because the manufacturer obtains the data directly from the patients (which is the case with many “wearables”), the professional code of conduct of the medical associations and the Declaration of Helsinki noted above do not apply. However, please consider the note with question 6.
In addition, the physicians employed by the manufacturer to perform the investigation are also subject to the responsibilities of their professional code of conduct, provided they are members of a German physician association.
The pertinent data protection regulations (in particular the GDPR and BDSG) apply independently of and in addition to the professional code of conduct.
Accordingly, manufacturers must observe the principles of data processing such as limiting the purpose and minimizing the data and the processing time (see GDPR Art. 5 paragraph c). They must also comply with the responsibilities as “controllers” and, for example, ensure technical and organizational measures to protect the data.
Figure 6: Does an ethics committee have to be involved?
Answer: As a rule, yes, but not for approval
Reason: As stated in the answer to the last question, physicians must consult with the ethics committee in the above cases in accordance with the professional code of conduct of their respective physician association. However, they are not required to obtain approval from an ethics committee under the Medical Device Regulation
This responsibility also applies to physicians employed by the manufacturer to perform the investigation, provided that they are members of a German physican association.
The views of the Permanent Working Party of Research Ethics Committees in Germany were published on the BfArM website. Unfortunately, they can no longer be accessed. The following chart is based on the content published at that time.
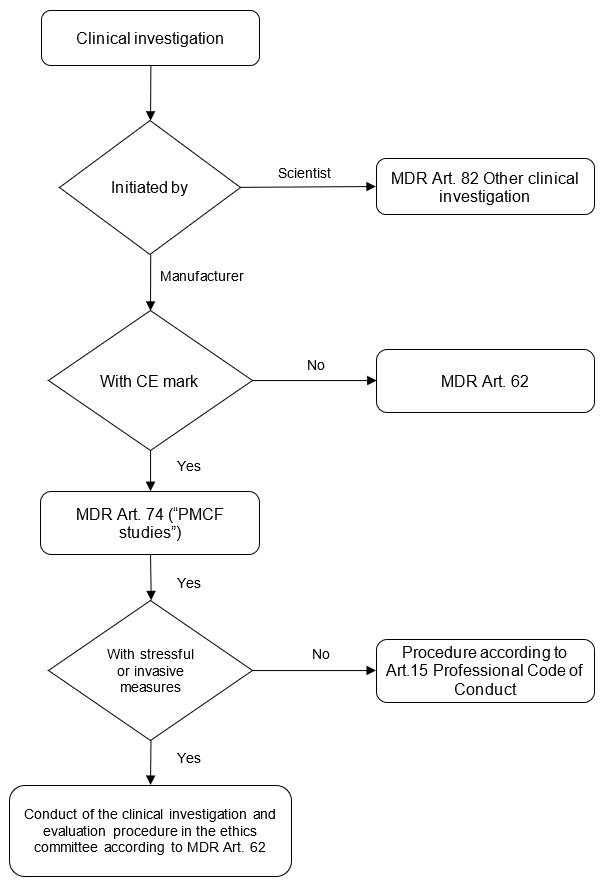
Figure 2: Clinical investigation flowchart
These presentations come to the same conclusion: When collecting PMS data, the following applies under the above conditions:
- The clinical investigation requirements, in particular MDR Art. 62, only apply to a limited extent.
- This is not an “other clinical investigation.”
- Approval is not required from the competent federal authority.
- If physicians are involved, they must comply with the professional code of conduct of their medical associations. However, there is no need for ethics committee approval.
Caution!
Please note that the Declaration of Helsinki also applies to persons other than physicians: The WMA encourages others involved in medical research on human participants to adopt these principles.
Figure 7: Does ISO 14155 apply?
Answer: Yes, but not all parts
Reason: ISO 14155 applies to clinical investigations. Its scope explicitly includes “clinical observational studies.” ISO 14155 imposes lower requirements on these observational studies.
Note
The objectives pursued by the legislator can also be derived from the
In Vitro Diagnostic Medical Devices Regulation (IVDR). It allows “experience gained by routine diagnostic testing” to be used to demonstrate the clinical performance of an IVD. For these data, too, the EU regulation requires that the ethical and regulatory principles be met. However, IVDs are not subject to ISO 14155, but ISO 20916. The latter describes good trial practice for “Clinical performance studies using specimens from human subjects.”
Figure 8: Does the MDR establish the legal requirement for GDPR-compliant data processing at the manufacturer?
Answer: No
Reason: GDPR Art. 6 (c) does allow data processing if “processing is necessary for compliance with a legal obligation to which the controller is subject.” However, there is no legal regulation under Member State or Union law meeting the requirements of GDPR Article 6 (3) that creates the authority to process data for the purpose of PMS or PMCF.
In particular, this does not follow from the definition of post-marketing surveillance (MDR Art. 2, No. 60), the concept of clinical data (MDR Art. 2, No. 48) or MDR Art. 61, Art. 83, Annex III and XIV Part B, the regulations of the MPDG and also not from the concept of data processing (GDPR Art. 4 No. 2).
In addition, manufacturers must keep in mind that clinical data and post-market data are data concerning health in the sense of GDPR Art. 4 No. 15. They can only be processed under the conditions specified in GDPR Art. 9 (2). Letter i) is particularly relevant here:
“processing is necessary for reasons of public interest in the area of public health, such as protecting against serious cross-border threats to health or ensuring high standards of quality and safety of health care and of medicinal products or medical devices, on the basis of Union or Member State law which provides for suitable and specific measures to safeguard the rights and freedoms of the data subject, in particular professional secrecy [...]”
However, there is no corresponding legal regulation under Member State or Union law establishing an authority to process data pursuant to GDPR Art. 9 (2) (i) for the purpose of PMCF.
4. Concrete statements
Here we collect statements from ethics committees, authorities, notified bodies, and manufacturers.
a) Assessment of a state ethics committee
The following assessment is from December 2022:
If the medical device to be tested bears a CE marking and no additional stressful or invasive measures take place within the scope of the study, nor is a study plan followed, it is another study according to § 15 of the BO for physicians, for which professional legal advice by the ethics committee at the Baden-Württemberg State Medical Association is necessary/possible only under the following conditions:
- It is a research project,
- in which a physician participates and
- in which the psychological or physical integrity of a human being is interfered with, or body materials or data are used that can be assigned to a specific human being.
Provided that the data are collected anonymously (i.e., without the possibility of attributing them to a specific individual) and otherwise do not interfere with the psychological or bodily integrity of an individual (esp., through investigations), the research project is not subject to professional review by the ethics committee.
Note: This article is based on the assumption that personal data are collected.
5. Conclusion & summary
In most cases, approval by an ethics committee is not required when collecting patient-related PMS data, but consultation with the ethics committees of physician associations or universities is required.
While this consultation is not legally binding, many (highly ranked) journals, however, do require ethics committee approval for publication. Therefore, if their advice were not followed, this requirement would not be met.
Most consultations take place within 3 to 4 weeks.
Thus, the hurdle posed by this consultation should not prompt manufacturers to fail to comply with the requirement to collect and analyze post-market data (particularly PMCF data). On the contrary, manufacturers should regard the consultation as an aid to complying with data protection requirements.
The appeal to the decision-makers at authorities, notified bodies and ethics committees is:
- Focus on the original meaning of the regulations.
- Do not overinterpret regulatory requirements. Feel free to follow the BfArM recommendations (“Neither the MDR nor the MPDG specify requirements for PMCF studies. (Meaning of the last sentence in MDR Article 74 (1): not generally applicable)”).
- Help manufacturers collect PMS data without unnecessary hurdles. Because if they refrain from doing so out of fear of you, you will achieve the opposite of what the regulation intends to accomplish: Greater patient safety.
The Johner Institute supports medical device manufacturers in
- creating and reviewing PMS and PMCF plans,
- the automated collection of post-market data,
- clinical evaluations and performance assessments.
Change History
2023-01-24: Statement of an ethics committee added

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.