MDCG 2020-6: Clinical Data Requirements for Legacy Devices
Currently, manufacturers of legacy devices can keep their devices on the market without demonstrating conformity under Regulation 2017/745 on Medical Devices (MDR). However, manufacturers will have to provide the necessary clinical data by the end of the transitional period at the latest.
However, the requirements for clinical data that can be used to demonstrate conformity have increased enormously under the MDR. Furthermore, the MDR is often vague with regard to what the precise requirements are.
The guidance document MDCG 2020-6“Regulation (EU) 2017/745: Clinical evidence needed for medical devices previously CE marked under Directives 93/42/EEC or 90/385/EEC – A guide for manufacturers and notified bodies” aims to rectify the situation. It gives recommendations on the requirements clinical data must meet in order to demonstrate a legacy device's conformity with the MDR.
Manufacturers of legacy devices should use the transitional period to collect the required clinical data, following this guide, if necessary, in good time to ensure they don’t have to take their device off the market.
The following article will make your work easier by:
- Summarizing the recommendations of MDCG 2020-6
- Giving tips on the best way to collect the required clinical data for your legacy device
1. Scope of MDCG 2020-6
a) “Legacy devices”: devices with MDD/AIMDD certificate
MDCG 2020-6 refers to what it calls “legacy devices.” These are existing devices that have already been placed on the market under EU Directive 93/42/EEC on Medical Devices (MDD) or Directive 90/385/EEC on Active Implantable Medical Devices (AIMDD) before the MDR came into force.
Definition: legacy device
"‘legacy devices’: this is considered to include all devices previously CE markedunder the European Medical Devices Directive 93/42/EEC (MDD) orActiveImplantable Medical Devices Directive 90/385/EEC (AIMDD)”
MDCG 2020-6 provides guidance for manufacturers and notified bodies on how to collect and analyze clinical data related to these legacy devices.
These data will later be important for the conformity assessment procedure according to the MDR.
b) Clinical data for the conformity assessment procedure according to the MDR
The entry into effect of the MDR (May 2021) triggered a transitional period for legacy devices; at the end of this transitional period, they will have to go through a conformity assessment procedure under the MDR. The end of this transitional period is May 26, 2024 at the latest.
Clinical data is needed to:
- Confirm compliance with the applicable general safety and performance requirements according to MDR Annex I
- Evaluate undesirable side effects and the acceptability of the benefit-risk ratio
Even during the transitional period, the MDR's requirements for post-market surveillance apply to manufacturers. As part of their post-market surveillance (PMS), they must proactively collect both data on the safety and performance of legacy devices and scientific data. These results are incorporated into the clinical evaluation, which is in turn essential for demonstrating conformity.
All pre-market and post-market data on the device can be used to demonstrate conformity with the applicable general safety and performance requirements in the MDR.
Therefore, manufacturers of legacy devices, in particular, should make sure they take a look at MDCG 2020-6's recommendations for collecting and evaluating this data.
Further information
You can learn more about the clinical evaluation in our overview article.
You can learn more about the MDR’s transitional periods in a separate article.
c) Who is MDCG 2020-6 particularly interesting for?
This means MDCG 2020-6 is particularly interesting for:
- Manufacturers of legacy devices
They can use the transitional period before they have to renew their MDR certificate to collect enough clinical data about their device.
- Manufacturers developing a new device
MDCG 2020-6 provides definitions for several terms that are not explicitly defined in Article 2 of the MDR but which are very important with regard to the evaluation of the benefit-risk ratio and the conclusions of the clinical evaluation (e.g., indication, state of the art, level of clinical evidence).
2. How MDCG 2020-6 helps with the MDR conformity assessment procedure
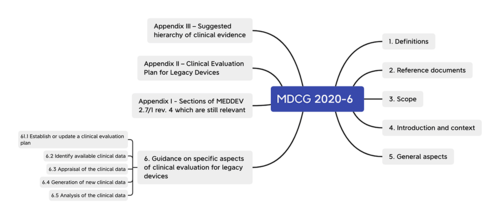
MDCG 2020-6 clarifies and amends some ambiguities in the MDR regarding clinical data. These include in particular:
- Definitions
In section 1.2, MDCG 2020-6 provides definitions of terms that are not defined in the MDR. These include “legacy device,” “well-established technology,” “scientific validity,” “sufficient clinical evidence,” “state of the art,” “intended use,” “indication,” and “similar device.”
- Clinical evaluation
In its “General aspects” section, MDCG 2020-6 highlights several points worth noting with regard to the clinical evaluation. Particular reference is made to the MEDDEV 2.12/2 guidance on PMCF studies in this section.
- Exemptions from the use of clinical data for non-implantable and Class III devices
Art. 61 MDR specifies some exceptions where clinical data do not have to be used to demonstrate conformity. MDCG 2020-6 clarifies when these apply and when they do not.
- Clinical evaluation plan
In Annex XIV, Part A, 1(a), the MDR specifies requirements for the clinical evaluation plan (CEP). MDCG 2020-6 recommends additional aspects that should be considered in addition to the requirements in the MDR.
- Clinical data appraisal, analysis and generation
MDCG 2020-6 gives recommendations on what to consider when appraising, analyzing and generating clinical data.
- Sections of MEDDEV 2.7/1 rev. 4 that are still relevant under the MDR
MEDDEV 2.7/1 rev. 4 actually refers to the now obsolete directives. However, there are sections that still apply under the MDR. These are listed.
- Hierarchy of clinical evidence
MDCG 2020-6 also provides a recommended order or hierarchy of clinical evidence for confirming a legacy device’s conformity with the MDR.
3. Recommendations and explanations from MDCG 2020-6
a) Definitions
In section 1.2, MDCG 2020-6 adds definitions that are missing from the MDR.
Legacy devices
“All devices previously CE marked under the European Medical Devices Directive 93/42/EEC (MDD) or Active Implantable Medical Devices Directive 90/385/EEC (AIMDD).”
Well-established technology
Although the MDR uses the term “well-established technology” in several articles (Art. 52(5), Art. 61(8)), it does not define the term.
In section 1.2, MDCG 2020-6 details the criteria that a well-established technology must meet:
- Relatively simple, common and stable designs with little evolution
- Their generic device group has well-known safety and has not been associated with safety issues in the past
- Well-known clinical performance characteristics and their generic device group are standard of care devices where there is little evolution in indications and the state of the art
- A long history on the market
Scientific validity
For scientific validity, MDCG 2020-6 emphasizes that clinical evaluations must follow a “defined and methodologically sound procedure.” The document mentions some criteria such as:
- Adequacy of study design and controls for bias
- Appropriateness and relevance of research questions
- Adequacy of sample sizes and statistical analyses
- Completeness of data
- Adequacy of follow up period
- Appropriateness of conclusions on the basis of objective evidence
It also makes reference to section 9.3.1 of MEDDEV 2.7/1 rev. 4, which is still valid under the MDR.
Sufficient clinical evidence
“Sufficient clinical evidence” is required but not defined in the MDR. MDCG 2020-6 rectifies this and defines “sufficient clinical evidence” as:
“the present result of the qualified assessment which has reached the conclusion that the device is safe and achieves the intended benefits”
State of the art
For this definition, the guideline borrows the definition given in IMDRF/GRRP WG/N47:
“Developed stage of current technical capability and/or accepted clinical practice in regard to products, processes and patient management, based on the relevant consolidated findings of science, technology and experience.”
Intended use
For this definition, the document merely states that “the MDR defines ‘intended purpose’, but not ‘intended use’. ‘Intended use’ should be considered to have the same meaning as ‘intended purpose’."
Indication
Indication is defined as “the clinical condition that is to be diagnosed, prevented, monitored, treated, alleviated, compensated for, replaced, modified or controlled by the medical device. It should be distinguished from ‘intended purpose/intended use’, which describes the effect of a device.”
Similar device
“Devices belonging to the same generic device group. The MDR defines this as a set of devices having the same or similar intended purposes or a commonality of technology allowing them to be classified in a generic manner not reflecting specific characteristics.”
c) Points of interest for clinical evaluations
According to MDCG 2020-6, “the MDR reinforces a number of important factors which are relevant to clinical evaluation”:
- Consideration of available alternative treatment options is required for the confirmation of the acceptability of the benefit-risk ratio.
- The acceptability of the benefit-risk ratio must be based upon clinical data providing sufficient clinical evidence including where applicable relevant data from post-market surveillance (PMS)
- The level of clinical evidence must be specified and justified by the manufacturer, taking the characteristics of the device and the intended purpose into account
- The incorporation of post-market surveillance (PMS) data, in particular the post-market clinical follow-up (PMCF) data, into the process of clinical evaluation
- Manufacturers are required to establish a post-market surveillance plan in accordance with Annex III of the MDR.
MDCG 2020-6 emphasizes the importance of post-market clinical follow-up (PMCF) data in particular.
Post-market clinical follow-up (PMCF) data involves the systematic collection of clinical data. The aim of the PMCF is to update the clinical evaluation.
Definition: Post-market clinical follow-up (PMCF)
“Continuous process that updates the clinical evaluation referred to in Article 61 and Part A of this Annex”
Source: MDR
d) Exemptions from the use of clinical data
Art. 61(10) of the MDR states that in cases where “the demonstration of conformity with general safety and performance requirements based on clinical data is not deemed appropriate,” other evidence can be provided.
MDCG 2020-6 indicates that these exemptions do not apply for class III or implantable devices.
In principle, any such exception must be justified “based on the results of the manufacturer's risk management and on consideration of the specifics of the interaction between the device and the human body, the clinical performance intended and the claims of the manufacturer.”
e) Establishing or updating a clinical evaluation plan
In Annex XIV, Part A, 1(a), the MDR specifies requirements for the clinical evaluation plan (CEP). In addition to the requirements specified in the MDR, section 6.1, MDCG 2020-6 recommends considering the following:
Minimum requirement according to MDR, Annex XIV, part A, 1(a) | Explanation in MDCG 2020-6 |
(1) Identification of the relevant general safety and performance requirements (GSPRs) | “The manufacturer should conduct an analysis with respect to the GSPRs of the MDR, to determine if additional data to support the clinical evidence are required to meet additional MDR requirements.” |
(2) and (3) Specification of the intended purpose, target groups, indications, contraindications | “Inputs for the clinical evaluation plan are in line with the device’s ‘label, instructions for use, promotional or sales materials or statements’” |
(4) Detailed description of intended clinical benefits with relevant and specified clinical outcome parameters | Additional information with respect to the quantification of benefits and determination of relevant clinical benefit outcome parameters with reference to MEDDEV 2.7/1 rev. 4 Annex A7.2 sections b and c. |
(5 – 7) Specification of qualitative and quantitative aspects of clinical safety and performance | Justification of the “level of clinical evidence”: “As an outcome of this step, it should be possible to conclude if the device is one which has a clearly positive benefit–risk determination.” |
(6) “determine…. the acceptability of the benefit-risk ratio for the various indications and for the intended purpose or purposes of the device”
| “Need to be based on the state of the art in medicine” (MEDDEV 2.7/1 rev. 4, section 8.2) |
f) Appraising, analyzing and generation of clinical data
In section 6.3, MDCG 2020-6 provides recommendations for the appraisal and analysis of existing and new data.
Identifying clinical data
Both pre-market and post-market data can be used to demonstrate conformity. These data sources include:
- State of the art
- Clinical data from similar devices
- Usability tests or simulated use testing
A summary of data sources is provided in Appendix III of MDCG 2020-6.
PMCF studies
PMCF studies may be used in addition to non-clinical data (Article 61(10) MDR) to confirm safety and performance.
Analysis of clinical data
When analyzing existing data to demonstrate conformity, MDCG 2020-6 differentiates between direct clinical benefits and indirect clinical benefits. The document understands a direct clinical benefit as one resulting from a medical device fulfilling a direct therapeutic or diagnostic function.
Medical devices with an indirect clinical benefit do not perform a direct therapeutic or diagnostic function. The clinical benefit is only indirect. The document gives the example of guidewires that “assist other medical devices in achieving their intended purpose, without having a direct therapeutic or diagnostic function themselves.”
- Direct clinical benefit
Must be demonstrated by clinical data. - Indirect clinical benefit
To demonstrate indirect clinical benefits, manufacturers can also use:- Pre-clinical/bench test data
- Data from the use of the device (e.g., from an insurance database)
- Data from another device that is used with the subject device (e.g., data from a stent used to justify the safety and performance of a guidewire).
Risks
The MDR requires manufacturers to have a risk management system. ISO 14971 provides guidance on how to set up and operate one.
The extent to which this risk management requires new clinical data to be generated depends on whether the likelihood and severity of a particular harm or the effectiveness of a risk control measure requires this.
A decision as to whether new clinical data have to be collected for risk management purposes should be one of the outputs of the clinical evaluation.
Benefit-risk determination
According to the MDR, the benefit of a medical device must always be proportionate to the risk that the device poses to patients. To achieve this, manufacturers must consider the following when collecting data:
- The state of the art
- Alternative treatment options
To determine these, manufacturers should, for example, refer to previously published clinical studies and treatment guidelines.
Lack of clinical data
If there is a lack of clinical data for a legacy device, MDCG 2020-6 recommends:
- Narrowing the intended purpose of the device
- Performing PMCF studies prior to certification under the MDR
- For well-established technology, review/undertake PMCF activities. In exceptional cases, a quality management system and post-market surveillance data are sufficient, but sometimes not for implantable and class III devices (see above)
Equivalent devices
Data from equivalent devices can also be used as evidence. However, the equivalence must be demonstrated separately. Still, the MDR’s requirements for equivalence are so high that they can rarely be used to provide evidence.
Nevertheless, manufacturers can still use data from similar (not equivalent) devices.
- Data from similar devices may also be important to establish whether the device under evaluation and similar devices belong to the group of devices considered as “well established technologies” (WET)
- Data from similar devices may be used, for example, to demonstrate ubiquity of design, lack of novelty, known safety and performance profile of a generic group of devices, etc.
Further information
You can learn more about the requirements for equivalent devices for the clinical evaluation in the article “MDCG 2020-5:The End of the Equivalence Route for Clinical Evaluations?”.
g) Sections of MEDDEV 2.7/1 rev. 4 that are still relevant under the MDR
Although MEDDEV 2.7/1 rev. 4 was not created for the MDR, parts of the document are still applicable under it. MEDDEV 2.7/1 rev. 4 provides specific instructions on how to structure and carry out the clinical evaluation.
MDCG 2020-6 specifically lists which sections of MEDDEV 2.7/1 rev. 4 are still applicable to the MDR.
- 6.4. Who should perform the clinical evaluation?
- 8. Identification of pertinent data (Stage 1)
- 9. Appraisal of pertinent data (Stage 2)
- 10. Analysis of the clinical data (Stage 3). This chapter includes references to the MDD, MDR requirements should be used instead
- A3. Device description - typical contents
- A4. Sources of literature
- A5. Literature search and literature review protocol, key elements
- A6. Appraisal of clinical data - examples of studies that lack scientific validity for demonstration of adequate clinical performance and/or clinical safety
- A7.2. Conformity assessment with requirement on acceptable benefit/risk profile
- A7.3. Conformity assessment with requirement on performance
- A7.4. Conformity assessment with requirements on acceptability of undesirable side-effects
- A10. Proposed checklist for the release of the clinical evaluation report.
h) Hierarchy of clinical evidence
MDCG 2020-6 recommends the following hierarchy for clinical and non-clinical data sources that manufacturers can use to demonstrate the conformity of their legacy device:
- Results of high-quality clinical investigations covering all device variants, indications, patient populations, duration of treatment effect, etc.
- Results of high-quality clinical investigations with some gaps
- Outcomes from high-quality clinical data collection systems such as registries
- Outcomes from studies with potential methodological flaws but where data can still be quantified and acceptability justified
- Equivalence data (reliable / quantifiable)
- Evaluation of state of the art, including evaluation of clinical data from similar devices
- Complaints and vigilance data; curated data
- Proactive PMS data, such as that derived from surveys
- Individual case reports on the subject device
- Compliance to non-clinical elements of common specifications considered relevant to device safety and performance
- Simulated use / animal / cadaveric testing involving healthcare professionals or other end users
- Pre-clinical and bench testing / compliance to standards
Note
Class III legacy devices and implantable legacy devices that are not well-established technologies should have sufficient clinical data as a minimum at level 4, namely through an assessment of cumulative evidence from additional sources (5-12). Reliance solely on complaints and vigilance is not sufficient.
4. What manufacturers should do now
a) Make the most of the transitional periods
Manufacturers of legacy devices should use the transitional period until they need to demonstrate compliance with the MDR. They should collect sufficient clinical data about their device.
b) Perform a gap analysis
Analyze the existing clinical data with regard to clinical evidence required for MDR conformity.
Perform a gap analysis during the clinical evaluation to identify any lack of clinical data.
- Not every device automatically needs a clinical investigation or PMCF study. Whether and in what form clinical data are required at all or can be used as evidence should be identified at an early stage through a clinical evaluation.
- In general, there must be sufficient clinical evidence to confirm the safety, performance and acceptability of the benefit-risk ratio with regard to the state of the art for the legacy device before CE marking under the MDR.
c) Establish PMCF activities
When establishing PMCF activities, incorporate the results of the gap analysis into the clinical evaluation.
d) Check whether devices can (still) be considered equivalent
The MDR’s requirements with regard to equivalence criteria are very high and it may be the case that clinical data from another device can no longer be used for your device. Use the MDR criteria to check whether the devices are still equivalent.
5. Conclusion
MDCG 2020-6 provides important guidance on how clinical data for legacy devices should be provided so it can be used to demonstrate conformity with the MDR. The document also clears up some ambiguities in the text of the MDR.
Manufacturers should look at MDCG 2020-6 early in the transitional period. Firstly, because the MDR requirements for post-market surveillance, including PMCF, apply even during the transitional period. Secondly because they should definitely not let the transitional period pass by without making the most of it.
The MDR has increased the requirements for clinical data, particularly for demonstrating equivalence. The activities required should therefore be established as quickly as possible so that an adequate set of data can be built up in good time.
If you have any questions regarding the clinical evaluation or the conformity assessment procedure, please contact our consultants. Simply use our contact form or send us an e-mail.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.