What Constitutes the Lifetime of a Medical Device in the EU?
Operating medical devices beyond the end of their lifetime can be dangerous and legally problematic. Therefore, manufacturers should precisely define the lifetime of every device. But, if you were put on the spot, would you be able to quickly explain how the lifetime is defined or what criteria are used to work it out?
If you couldn’t, you’re not alone: a lot of aspects regarding the lifetime of medical devices are unclear, especially at the European level.
Related terms such as “shelf life” and “operating time” often cause additional confusion – as does the fact that the lifetime does not necessarily have to be a “duration,” other factors can also play a role.
Avoid problems with regulatory authorities and patient-related costs, or even damages, and find out in this article:
- Why working out the lifetime is important
- How to define and establish the lifetime
- How lifetime differs from other concepts such as end of service or operating time
- Which (regulatory) requirements you must comply with as a manufacturer and operator
1. Why the lifetime and its definition are important
The lifetime of a medical device is the period during which the device is safe to use and performs as intended. Naturally, a device's properties change over time, for example, due to aging or wear. Major problems can arise if these changes mean that safety and/or performance are no longer guaranteed. For example:
- A dialysis machine's fluid balancing becomes inaccurate over time as the sealing materials age, resulting in fluid losses.
- Plastics become brittle and lose their fracture strength.
- Electronic components can malfunction after a lot of switching operations.
The lifetime of medical devices is important for both manufacturers and operators.
a) Importance for manufacturers
Manufacturers have to know the lifetime when formulating the intended purpose.
- They need the lifetime for the risk management process (Annex I MDR, ISO 14971), where they need to analyze whether the device is safe and performs as intended over its entire lifetime.
- The lifetime correlates with the total number of uses. Both affect the risk acceptance matrix. The class of the least probable harm (usually the bottom row) should have probabilities that mean they shouldn’t be expected to occur during all uses over the complete lifetime of the device.
- The intended lifetime conditions the selection of components, materials and technologies even during development.
- The lifetime and necessary service intervals are closely linked to one another. The manufacturer documents these service intervals in the accompanying information.
b) Importance for operators
Operators also have to act at the end of the device's specified lifetime (e.g., by taking the device out of service). Operators often have a hard time with this because the device seems to be working flawlessly. They cannot see how the safety or performance is compromised. This is probably also the reason many operators struggle with decommissioning.
It is even more difficult if the manufacturer has not specified the lifetime in the accompanying documentation. In this case, how is the operator supposed to know when safety and performance are no longer guaranteed?
Even if operators think their device is still working perfectly: the lifetime specified by the manufacturer takes precedence.
c) Importance for customers
The lifetime of a device is also an important factor for customers. Indeed, in a lot of cases, it is one of the factors taken into consideration when deciding whether to purchase a device. If a purchaser at a hospital purchaser has a choice between two otherwise similar devices, they will choose the one with the longer lifetime.
2. The complicated definition of lifetime
As important as it is for manufacturers, operators, customers and authorities to know what the lifetime of a medical device is, it is just as difficult to precisely define (and thus establish) what constitutes the “lifetime.” Legal regulations mostly remain vague, and a lot of terms are used synonymously with “lifetime.”
a) MDR
EU Regulation 2017/745 (MDR) does not explicitly define lifetime. However, in Annex I, it does summarize the general safety and performance requirements and indirectly and incidentally define the term “lifetime”:
The characteristics and performance of a device shall not be adversely affected to such a degree that the health or safety of the patient or the user and, where applicable, of other persons are compromisedduring the lifetime of the device, as indicated by the manufacturer, when the device is subjected to the stresses which can occur during normal conditions of use and has been properly maintained in accordance with the manufacturer’s instructions.
Source: MDR Annex I, Chapter I No. 6
“As indicated by the manufacturer” refers to “the lifetime of the device” and this enables us to understand the requirement for the manufacturer to specify the lifetime of the device.
b) IMDRF
While the MDR does not explicitly define the term lifetime, there is a more detailed explanation of the concept in the “Essential Principles of Safety and Performance ” document from the International Medical Device Regulators Forum (IMDRF). According to this explanation, the “expected lifetime” is:
Definition: “Expected lifetime” according to the IMDRF
“Time period specified by the manufacturer during which the medical device or IVD medical device is expected to maintain safe and effective use.”
Source: IMDRF Essential Principles of Safety and Performance
Section 2.3 of the old IMDRF document “GHTF SG2 N21 R8 May 1999” made the “service life”, as it called it, dependent on the intended duration of use:
Service life is defined as: the time or usage that a device is intended to remain functional after it is manufactured, placed into use, and maintained as specified.
Source: GHTF SG2 N21 R8 May 1999 Section 2.3
c) Active medical devices: IEC 60601-1
For active medical devices, IEC 60601-1 defines the “expected service life” as the:
time period specified by the manufacturer during which the ME equipment or ME system is expected to remain safe for use (i.e., maintain basic safety and essential performance)
Source: IEC 60601-1
d) How you should deal with the different definitions
As evidence of compliance with the MDR's general safety and performance requirements as defined in MDR Annex I, paragraph 6 has to be provided in the device's technical documentation, we recommend documenting your definition of the term “lifetime” there as well.
One manufacturer might think of the lifetime as the number of uses (“300 uses”), another as a time period (“up to 5 years after commissioning”) or up to a specific date (“until 12/2025”). Combinations are also possible: 5 years or 1,000 treatments, whichever comes first.
Note for manufacturers
It is important that the lifetime should not, as a general rule, be the result of device development. Instead, it should be determined prior to device development and be used as a “design input.”
However, there are exceptions to this rule: for example, in the case of reagents, where manufacturers may not be able to definitively define the lifetime until after verification and validation.
3. What the lifetime is NOT (and what you nevertheless need to pay attention to)
It is also important to know which terms, although often used in connection with lifetime, should not be confused with it.
a) Best before date
Most of us are familiar with the idea of a "best before date” from the food industry. But this term is not (or is no longer) used in the medical device sector. In contrast to foodstuffs, where the best before date is more of a guide, the shelf life and lifetime of a medical device are non-negotiable.
This means that after the end of the specified period or when a specified expiry date has been reached (see ISO 15223: “use-by-date”), safety and/or performance are no longer guaranteed and, therefore, the device may no longer be used.
b) Stability, including shelf life
In ZLG Document 3.3 A 5 (Shelf life – Evaluation of the defined shelf life) shelf life is defined as:
Shelf life before the first use of the device as specified by the manufacturer, provided the storage and transport conditions are complied with.
The shelf life ends with the first use of a device, for example, when its packaging is opened (e.g., dental filling materials for several portions across multiple uses).
The shelf life is particularly important for sterile devices that must remain sterile until first use, including during transport and storage. In this case, the manufacturer specifies for how long sterility is assured under specified conditions.
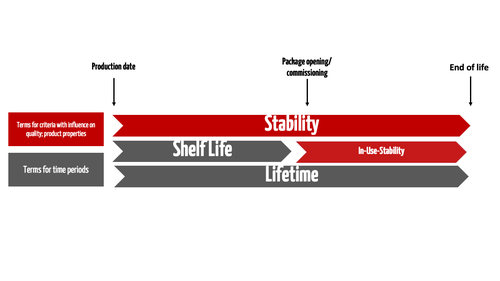
The term “stability” is generally used to describe a device characteristic. The manufacturer must provide evidence of stability for both the shelf life and the lifetime (see Fig. 1).
Since a medical device's safety and performance can also be impaired after the device's first use, from this point until the end of the device's lifetime, we use the term in-use stability (e.g., for dental filling materials).
A device's lifetime starts at the time of production and, therefore, includes the shelf life.
First summary
- Lifetime (or service life): Total period of time from production to the end of a device's life during which the device is safe and performs as intended
- Shelf life: Maximum period of time between production and commissioning/use during which the device is still safe and performs as intended
- Stability: Device property indicating that the device remains safe and performs as intended throughout the device’s life cycle
- In-use stability: Time between commissioning/opening of packaging and end of life
Examples
- An implant may have a lifetime of 15 years (including time in the human body), but a shelf life of only 2 years in terms of stability in its original packaging
- A substance-based medical device may be stored in its original packaging for three years (shelf life) but have to be used within 12 months of being opened (in-use stability)
Stability of IVD reagents
The stability of reagent devices, including calibration and control materials for in vitro diagnostic medical devices, is particularly important. They are generally very susceptible to environmental influences such as light, temperature or vibration.
The stability testing must be part of the technical documentation according to Annex II, 6.3 of the IVDR.
- Relevant standard: ISO 23640 “Evaluation of stability of in vitro diagnostic reagents”
- Relevant guidance document: CLSI guidance document EP25-A
ISO 23640 is expected to be harmonized with the IVDR. The standard is supplemented by CLSI guidance document EP25-A. Both require real-time testing under realistic conditions. However, they also allow for “accelerated stability testing” to enable an initial estimate of the stability and shelf life of a device.
c) End of service
Although the terms are similar, the end of service is not aligned with the lifetime.
Definition: End of service
The end of service is the end of the availability of spare parts and the retrofittability of the device.
This period is determined by the manufacturer or the service organization and is not defined from a regulatory perspective unless the manufacturer cancels services before the end of the device's lifetime. In this case, the medical device can no longer be operated safely and perform as intended due to a lack of maintenance and repair and may have to be taken out of operation or withdrawn from the market by the operator.
d) Operating period
The operating period is the name given to the period of active operation itself, i.e., the period when the device is actually used. This is not the same as the lifetime, as the lifetime does not necessarily depend on actual use.
Example:
A dialysis machine has a lifetime of ten years. Even if the device is only used for half an hour during these ten years, the lifetime ends at the end of these ten years.
The operating period, therefore, depends less on the device than it does on the area of use and its users. It often correlates to a certain extent with the lifetime, but not always because downtimes, storage and transport also cause aging or other effects.
Because the age-related and the use-related lifetime can be different, as mentioned above in some cases both are specified: 5 years or 1000 treatments, whichever comes first.
e) End of life
The IMDRF's document on cybersecurity defines the end of life as:
End of Life (EOL): Life cycle stage of a product starting when the manufacturer no longer sells the product beyond their useful life as defined by the manufacturer and the product has gone through a formal EOL process including notification to users.
So, it is the time at which a manufacturer ceases to market a device. Therefore, “end of life” refers to a product line not, as in the case of the lifetime, an individual device!
However, the US FDA uses the term “end of life” (EOL) differently. In 21 CFR 803.3, subparagraph (f) states:
Expected life of a device means the time that a device is expected to remain functional after it is placed into use. Certain implanted devices have specified “end of life” (EOL) dates. Other devices are not labeled as to their respective EOL, but are expected to remain operational through activities such as maintenance, repairs, or upgrades, for an estimated period of time.
Careful!
This means that, in the USA, “end of life” and “lifetime” are equivalent.
f) How you should deal with the different terms
Our tips:
- Use the terms used in the regulatory requirements
- List the terms in your documentation. Explicitly provide definitions again in a glossary
- Provide a reference to the origin of the definition in your glossary or state that it is your own definition
- Be aware that there may be several aging and lifetime-related aspects that are all relevant for your device. Corresponding proof is required for each individual aspect
4. Determining the lifetime
a) Determining the lifetime of physical devices
General criteria for determining the lifetime
Unfortunately, legal regulations such as the MDR and IVDR do not provide any direct guidance on how the lifetime of a medical device should be determined.
Therefore, when determining the lifetime of a physical device, manufacturers should use relevant standards as a guide. However, there is no standard that covers all cases.
The now-withdrawn TR/ISO 14969:2004, which still referred to the application of ISO 13495:2003, had a very helpful summary of how to define the lifetime of medical devices in section 7.1.3:
Decisions related to product lifetime can be made, in part, to control identified residual risks that can increase to unacceptable levels as the period of use of a medical device is extended.
The standard also described other factors that should be considered when defining the lifetime:
a) shelf life of the medical device
b) expiry date for medical devices or components which are subject to degradation over time
c) number of cycles or periods of use of the medical device, based on life testing of the medical device
d) anticipated material degradation
e) stability of packaging material
f) for implantable devices, the residual risk that results from the entire period of residence of the device inside the patient’s body
g) for sterile medical devices, the ability to maintain sterility
h) organization’s ability/willingness or contractual or regulatory obligation to support service
i) spare parts cost and availability
j) legal considerations including liability
k) number of uses or operating hours
In addition, commercial software products that can model lifetime are also available.
Special standards for individual topics
There are up-to-date standards for specific topics. We recommend performing a detailed search of standards databases, e.g., iso.org or its full text search, to identify these standards.
Examples of specific standards are:
- ISO 11346:2014 Rubber, vulcanized or thermoplastic — Estimation of lifetime and maximum temperature of use
- ISO 17526:2003 Optics and optical instruments — Lasers and laser-related equipment — Lifetime of lasers
- ISO/IEC 16963:2017 Information technology — Digitally recorded media for information interchange and storage — Test method for the estimation of lifetime of optical disks for long-term data storage
There are also standards describing aging tests for various materials. Search for “accelerated aging test” using the standards database’s full-text search.
Examples of standards on aging tests:
- ISO 188:2011 Rubber, vulcanized or thermoplastic — Accelerated aging and heat resistance tests
- ISO 23640 Evaluation of stability of in vitro diagnostic reagents
NB!
When defining the lifetime, it is vital that the environmental conditions defined in the device's extended intended purpose, particularly the effects of temperature, moisture, air pressure and UV light, and the corresponding storage and transport conditions, are taken into consideration.
Maintenance
There may be opportunities to check aged components through regular tests prior to use or through self-testing by the device and thus to detect and counteract unsafe conditions or reduced device performance in good time.
Using information on aging and from risk management, maintenance strategies that will extend the specified lifetime can be developed.
b) Determining the lifetime of software devices
Software itself does not age. But the software's environment does change: new operating systems, new technologies, new communication protocols, etc.
A pure software product will therefore rarely have a lifetime longer than a few years without modifications. Five years would be a good rule of thumb, although it should be calculated based on risk.
Additional information
You can find out more in the article “Lifetime of software products”
C) Tips for determining product lifetime
There are hardly any guidelines in the west on how to determine product lifetime but a guidance document from China has compiled some information. We have taken the following tips from it to help you to determine your product lifetime:
Step 1: establishing the desired product lifetime in the requirements specification
Prior to or at the beginning of the development stage, manufacturers should determine the product lifetime.
Indications of how long this period should be can be derived, for example, from the following considerations:
- commercial considerations: comparison with competitors’ products; customer requirements; profitability, etc.
- technical considerations: feasibility and time until components wear out
- empirical data: empirical values from current market surveillance and test results
As one of the stakeholder requirements, the lifetime is a specification and not a result of development. Rather, the product management department puts forward a proposal regarding the lifetime along with the stakeholder requirements. Via risk management, technical data sheets or empirical tests, the development team assesses the lifetime proposal and confirms it, raises any issues or offers solutions (e.g. preventive maintenance, more expensive components, etc.).
Step 2: creating a system and component design
The product is designed with the previously established requirement specifications in mind. Here, among other things, suitable technology and product components need to be identified that meet the requirements necessary to achieve the lifetime and fulfill all other product requirements.
The design should demonstrably include the calculations and derivations for selecting or designing certain components. On the basis of the calculations, an auditor would decide whether or not certain tests need to be carried out. Good documentation also helps to limit the scope of the tests.
The following aspects should be included in the design documents:
- derivation of the lifetime
- list of critical components
- reliability calculations or results of simulations
- calculations of safety factors according to Table 21 of IEC 60601-1
- environmental conditions that affect the lifetime
- results of the risk assessment
- if necessary, test requirements to prove the lifetime
- comparison with reference designs
- standards that can be observed
Step 3: analyzing the risks associated with the product lifetime and implementing measures
In risk management, manufacturers must identify the risks to the product and components that are linked to safety and performance beyond the lifetime of the components.
Here, the following points are particularly to be considered:
- the product itself
e.g. friction between two movable parts, actions of force - the usage criteria
e.g. frequency and intensity of use, maintenance and repairs - the environment
e.g. transport, storage, operating environment (temperature/humidity, UV light) - cleaning and disinfection
- combinations of different influencing factor
Cleaning and disinfection
Cleaning and disinfection are factors that are frequently overlooked when determining the product lifetime. Use of the product may involve cleaning and disinfection. The cumulative effect of heating or drying procedures and chemical residues from such procedures can affect the product’s performance and should be assessed in terms of the effects on the product lifetime.
Different sterilization methods (steam sterilization, ethylene oxide sterilization, radio sterilization, etc.) and packaging methods can also have different effects on the product lifetime. Therefore, for example, the effects of the sterilization procedure (methods and parameters) on the product lifetime should be considered.
Ways of extending the lifetime
For each component, the materials and operating principles must support the required lifetime. If this is not possible, the following options can guarantee the component lifetime and minimize risks:
- option 1: maintenance
Manufacturers can allow for preventive replacement of components. In this case, the corresponding maintenance intervals must be included in the instructions for use (IFU). - option 2: test function
Alternatively (or additionally), manufacturers can implement a test function on the product that reliably detects component failure (or other safety and performance limitations). This is always an option if the test function makes the product safer. However, it also means that this option is not effective in the case of life-sustaining/life-support systems. If their performance is limited or they malfunction and the test function is triggered, it is already too late. - option 3: reducing the product lifetime
If neither of the other options is feasible, all that remains is for manufacturers to reduce the product lifetime and adapt it to the requirement specifications.
5. Regulatory requirements for the lifetime of medical devices
There are no legal regulations that exclusively and conclusively deal with the lifetime of medical devices. Instead, it is always individual aspects that are regulated. These include in particular:
- Full refurbishment and effect on lifetime
- Labeling of lifetime
- Quality management system and lifetime
- Communication of the lifetime
- Active medical devices and lifetime
a) Full refurbishment lets the lifetime start afresh
‘Fully refurbishing’, for the purposes of the definition of manufacturer, means the complete rebuilding of a device already placed on the market or put into service, or the making of a new device from used devices, to bring it into conformity with this Regulation, combined with the assignment of a new lifetime to the refurbished device;
Source: Art. 2(31) MDR
This means medical devices can be restored to a condition that is either the same as the original condition of a new device or to a condition for which a new lifetime has to be specified.
The manufacturer must decide and document the effect of the refurbishment individually in each case. Cleaning, disinfection and sterilization aspects may also have to be considered. ISO 17664 can provide some guidance in this regard.
Full refurbishment may also mean a medical device no longer has to be disposed of. Whether or not this makes financial sense is up to the operator or the manufacturer to decide.
Operators generally rely on the manufacturer’s expertise when deciding on the scope, methods and details of a full refurbishment. However, there is no legal obligation for them to provide advice, which can be a challenge for operators.
b) Lifetime must be specified in the accompanying documents
For implantable devices, the MDR states:
1. The manufacturer of an implantable device shall provide together with the device the following: […]
c) any information about the expected lifetime of the device and any necessary follow-up;
Source: Art. 18 MDR
This is the only place in European regulations that explicitly requires the lifetime of a device to be stated in the information accompanying a device. However, other regulations in the MDR refer to information regarding lifetime in the accompanying documents.
Annex I MDR, section 23.4: information in the instructions for use
k) the information needed to verify whether the device is properly installed and is ready to perform safely and as intended by the manufacturer, together with, where relevant:
[…]
— information on any necessary calibration to ensure that the device operates properly and safely during its intended lifetime, and[...]
This means that, in addition to the replacement of wear parts and regular inspection of medical devices during maintenance, calibrations, which the manufacturer must define, also play a role.
Annex VI MDR, Part B: the UDI system
4.10. Devices that are reusable shall bear a UDI carrier on the device itself. The UDI carrier for reusable devices that require cleaning, disinfection, sterilisation or refurbishing between patient uses shall be permanent and readable after each process performed to make the device ready for the subsequent use throughout the intended lifetime of the device.
4.11. The UDI carrier shall be readable during normal use and throughout the intended lifetime of the device.
This section is about the labeling of devices but is more concerned with the interpretation of the labeling. The manufacturer must be able to demonstrate that the information plate with UDI is permanently affixed to the device, is always legible and withstands all external influences it is exposed to when the device is used as intended. The selection of materials for the label plays just as important a role as the printing ink and technique, light and temperature influences, fluids and humidity, vibrations and mechanical loads or the choice of appropriate disinfectants.
With regard to the permanent legibility of information plates, it must be ensured that the replacement of the device components the information plate is affixed to is technically and procedurally possible.
Have technicians in the field been informed of the situation? Can new information plates with a device's original data be generated on request? Can they be affixed in the field? How will they be verified in that case? It is these seemingly trivial special cases that rip holes in otherwise flawless regulatory compliance.
c) Lifetime conditions the PMS and PMCF
Article 83 MDR: post-market surveillance
The MDR establishes requirements for post-market surveillance (PMS):
2. The post-market surveillance system shall be suited to actively and systematically gathering, recording and analysing relevant data on the quality, performance and safety of a device throughout its entire lifetime, and to drawing the necessary conclusions and to determining, implementing and monitoring any preventive and corrective actions.
In this case, lifetime seems to refer not only to the individual device but to all devices on the market. This point is particularly important if the manufacturer has already discontinued the device (see “end of life”) but must continue to carry out post-market surveillance, until the last device on the market has reached the end of its defined lifetime to be exact. If it can be demonstrated that there are no devices still on the market, the post-market surveillance can also be ended.
Article 86 MDR: periodic safety update report
Throughout the lifetime of the device concerned, that PSUR shall set out: […]
This also applies to all devices on the market and describes the obligation to collect and evaluate specific information and to summarize it in a regular report.
Annex XIV, MDR: post-market clinical follow-up
The MDR also requires a post-market clinical follow-up (PMCF). This also depends on the lifetime:
5. When conducting PMCF, the manufacturer shall proactively collect and evaluate clinical data from the use in or on humans of a device which bears the CE marking and is placed on the market or put into service within its intended purpose as referred to in the relevant conformity assessment procedure, with the aim of confirming the safety and performance throughout the expected lifetime of the device, of ensuring the continued acceptability of identified risks and of detecting emerging risks on the basis of factual evidence.
As the lifetime is, by definition, the period of time during which a medical device can be operated safely and performs as intended, it is necessary to collect sufficient data throughout this period. This enables the manufacturer to demonstrate that all its assumptions and specifications were justified.
d) Communication of the lifetime
Users and operators must know from what point they may no longer operate the device because its safety and performance are no longer guaranteed. This alone makes it logically necessary for the manufacturer to communicate the lifetime.
But what are the mandatory legal requirements for this?
MDR
In fact, Article 18 (1)(c) of the MDR only requires a mandatory indication of lifetime in the case of implantable devices (see above).
IMDRF
Section 2.3 of the IMDRF document "GHTF SG2 N21 R8 May 1999" states:
The service life must be specified by the device manufacturer and included in the master record [technical file] or,where appropriate, the instructions for use (IFU).
“Where appropriate” can only be understood to mean that there may also be devices with a practically unlimited lifetime for which such information would make no sense. The same applies to sterile disposable products, for which the shelf life is more relevant than the lifetime since they are usually used immediately and then disposed of.
IEC 60601-1-11:2015
IEC 60601-1-11:2015 explicitly requires the expected service life to be specified in the documents accompanying medical electrical equipment for use in a domestic environment.
In general, it would be correct to say that, in most cases, a medical device's lifetime absolutely must be specified in the accompanying documents and, in some cases, it is even required by law or regulation. In any case, it must be included in the technical documentation.
e) Active medical devices according to IEC 60601
In IEC 60601-1, the lifetime is particularly relevant in connection with the term “single fault safe”.
Single fault safe is an important concept in IEC 60601-1. The standard defines it as:
Single fault safe
Characteristic of ME equipment or its parts whereby it remains free of unacceptable risk during its expected service life under single fault conditions.
Manufacturers can only comply with this requirement if they have defined the service life and can demonstrate it with empirical tests and risk management in an appropriate and transparent way.
Edition 3.1 of IEC 60601-1 makes the following plea to manufacturers:
In the accompanying documents, the manufacturer should provide information to allow the responsible organization to assess when the ME equipment is approaching the end of its lifetime. Such information should include the expected service life as determined by the manufacturer (e.g. in terms of years of service or number of uses) but could also include tests to be performed as part of preventive maintenance, or other criteria to allow the responsible organization to make an appropriate determination. The need for such information and the appropriate way to present it should be addressed as part of the risk management process.
f) Requirements in China
Manufacturers are currently getting some nasty surprises when trying to get their devices authorized in China. Lifetime plays such an important role there that there is even separate NMPA guidance for it. Its title, translated literally, is Guideline for the Technical Review of the Lifetime of Active Medical Devices (No. 23 of 2019)”.
When registering medical devices, applicants must provide evidence to support the lifetime of their device as specified in the accompanying documents.
The regulations describe in detail how the lifetime should be defined and how it is taken into account in risk management.
The requirements are basically the same as the regulatory requirements in Europe, but here, in contrast to China, it is much less likely to become a point of attack during the authorization process.
6. Conclusion
A medical device's safety and performance are only guaranteed during its specified lifetime. All aspects that affect risk must be taken into account when defining the lifetime. Unfortunately, a lot of manufacturers and operators take some liberties with the concept, in part because a longer lifetime can be economically advantageous and the definitions are fuzzy.
Terms such as lifetime, shelf life, end of service, operating time and end of life are not synonyms and they have to be understood precisely if you want to comply with the regulatory requirements.
In Europe, lifetime plays a major role in relevant documents, such as the risk management file and test documentation. IEC 60601-1 provides valuable information on how to implement this in practice.
Markets such as China also require the lifetime to be specified in the accompanying documentation, and also require information on how it was calculated as well as reliable proof in the technical documentation.
Manufacturers in Europe would therefore do well to specify the desired lifetime as a design input in the future. They should consider it an “essential design output” (as defined by 21 CFR 820.30d) to ensure closer attention is paid to the actual lifetime of their devices in the lifecycle process and also provide sufficient evidence to support their claims.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.