
Author:
Nadine Jurrmann
When can clinical investigations be avoided for devices with artificial intelligence?
For manufacturers, the answer to whether and when clinical investigations are necessary when using artificial intelligence in medical devices is relevant. After all, the duration and cost of bringing these devices to market depend on this.
The good news in advance: there are cases where manufacturers can avoid clinical investigations for devices with AI. This article explains which cases these are.
1. Summary for readers in a hurry
In most cases, manufacturers will not be able to demonstrate compliance with regulatory requirements (e.g., performance) based on published clinical data. That is because the equivalence of the devices or AI models used to generate the data cannot be adequately demonstrated.
However, it is possible to use retrospective analyses to avoid prospective clinical investigations for devices with AI.
2. The legal situation
The legal situations in the EU and the USA differ. In its guidance document on Clinical Decision Support Software, the FDA defines four criteria that a software application must meet to not be considered a medical device in the USA and not to be monitored by the FDA. However, these requirements are usually not met by AI-based software, for example, because the software is used for the analysis of images or signals.
Further information
Read more about the qualification of software as a medical device.
Furthermore, there are articles on the regulatory requirements for AI-based medical devices and the applications of AI in medicine.
The MDR (analogous to the IVDR) requires manufacturers to demonstrate devices' safety, performance, and clinical benefit as part of the clinical evaluation or performance evaluation of IVDs. As different as the MDR and IVDR requirements for clinical evidence may be for medical devices and IVDs, they are comparable for software. The MDCG describes the special features and similarities in the clinical evaluation or performance evaluation of medical device- or IVD software in its 2020 guidance document Guidance on Clinical Evaluation (MDR) / Performance Evaluation (IVDR) of Medical Device Software, which is well worth reading.
If insufficient clinical data is available, the manufacturer must generate it. As a rule, this means that a prospective clinical investigation must be conducted.
Notes
It is (almost) impossible to show clinical performance for an AI-based software based on literature data. This is almost only successful if one has written the publication oneself and thus conducted the study and the raw data are available. In this case, using the study data directly and saving the detour via the publication is usually easier.
Nevertheless, it is often strategically reasonable and possible to use already available datasets - provided that the datasets were not used for training and testing and that they are independent validation datasets with an appropriate statistically relevant number of cases for all subpopulations.
The MDR refers to clinical investigations, and the IVDR refers to clinical performance studies but not clinical trials. This article uses both terms synonymously.
3. The problem with equivalence
a) MDR requirements
Especially under the MDD, it was common practice to use clinical data from existing devices, thus avoiding the need for clinical investigations. The prerequisite was that the data came from equivalent devices. This also includes their technical equivalence. The required proof of technical equivalence explicitly also concerns the software algorithms:
Technical: the device is of similar design; is used under similar conditions of use; has similar specifications and properties including […] software algorithms […] and has similar principles of operation and critical performance requirements;
MDR, Annex XIV, 3. Paragraph, 1. Bullet
For class III medical devices and implantable devices, it becomes even more difficult to avoid clinical investigations:
[…] CLINICAL EVALUATION of class III and implantable devices (MDR) shall include data from a CLINICAL INVESTIGATION unless the conditions of Article 61(4), (5) or (6) of the MDR have been fulfilled.
MDCG 2020-1, Chapter 4.4
b) Technical equivalence in artificial intelligence
In the case of medical devices that use artificial intelligence and, in particular, machine learning, the models correspond to software algorithms (e.g., models such as the various forms of neural networks). These differ concerning:
- architecture of the models, e.g.,
- number of layers
- number of neurons per layer
- linkage of the layers
- activation functions (e.g., RELU)
- linking of submodels as for LSTM or LLM
- values of the fitted parameters (weight and bias)
The hyperparameters used to train the models affect the fitted parameters. However, their choice is not an aspect of technical equivalence. On the other hand, the "application conditions" are part of the technical equivalence.
So far, there is no specification of what the application conditions include in this context. Possible aspects are:
- technical environment such as hardware and operating system
- programming language and libraries used
- use and embedding of the model in the overall medical device
c) Demonstration of technical equivalence
Usually, manufacturers will not succeed in proving the equivalence of the "software algorithms" without comparing the software code of the equivalent product. As the MDCG states, this is also not necessary.
“It is the functional principle of the software algorithm, as well as the clinical performance(s) and intended purpose(s) of the software algorithm, that shall be considered when demonstrating the equivalence of a software algorithm. It is not reasonable to demand that equivalence is demonstrated for the software code, provided it has been developed in line with international standards for safe design and validation (IEC 62304, and IEC 82304-1) of medical device software.“
Comment in MDCG 2020-5, Chapter 3.1 (b)
For AI models, equivalence is not proven even with identical software code. This is because the software code (as part of the medical device) defines the AI model but not the values of the fitted parameters. These are determined by the training data and the type of training (e.g., hyperparameters). However, the software code for the training is not part of the medical device and is not covered by MDCG 2020-5.
MDCG 2020-5 requires that the "functional principle" and "clinical performance" (more on this later) be used in the equivalence comparison. For AI applications, the model architecture most closely corresponds to the "functional principle."
Attention
There is currently no consistent assessment by the authorities and notified bodies as to whether the same or similar model architecture is sufficient to demonstrate technical equivalence. Therefore, manufacturers should always determine and compare the "clinical performance" of both devices. These comparisons must be based on adequate scientific justification. Such a comparison will be difficult to make without access to the other model and/or the technical documentation of the other device.
If there is CE-marked AI software with the same intended purpose on the market, then comparing the clinical performance of the two MDSW products would be the objective of the needed clinical investigation.
The already approved AI with the same intended purpose would be the method of comparison, and the available validation data sets with both AI software devices would be analyzed, and the percent of agreement examined. If necessary, another reference method would be added - depending on the already approved AI software's quality and the clinical or performance evaluation strategy.
Sufficient clinical data are needed to demonstrate compliance with the essential safety and performance requirements. Clinical data can also be collected through other than clinical investigations (see next chapter).
4. Proof of conformity
a) Aspects/requirements to be proven
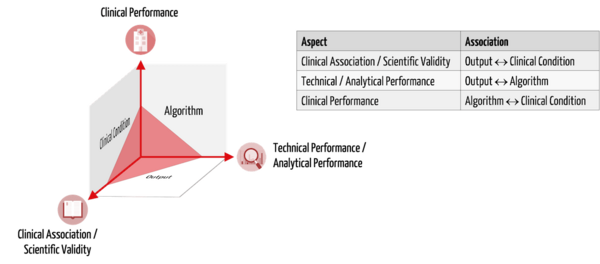
Manufacturers must use clinical data to demonstrate the safety, performance, and clinical benefit of their medical devices or IVDs. In the case of "Medical Device Software" (MDSW), this means, according to MDCG 2020-1, the demonstration of:
- clinical association/scientific validity
- technical performance/analytical performance
- clinical performance
Note
MDSW includes standalone software and software as part of a medical device or IVD when it directly serves its intended use. An example is the software of a defibrillator that detects a patient's heartbeat.
Table 1 presents these three aspects of clinical evidence using the example of AI-based medical device software that identifies tissue suspected of having cancer in CT images.
Aspect to be proven | Description | Example |
clinical association/scientific validity | There is a relationship between the software output and the physiological/clinical condition being tested. | The brightness values1) marked by the software actually describe cancerous tissue. |
technical performance/analytical performance | Given inputs reproducibly lead to correct outputs using the algorithm. | The software correctly and reproducibly marks the spots with certain brightness values in the images1). |
clinical performance | The intended use is achieved for all intended patients (with respective diseases) in the appropriate context of use by the intended users. | Intended users correctly detect cancer even in images from other CTs, patients in prone positions, concurrent tissue calcifications, etc. Metrics such as diagnostic sensitivity and specificity determine clinical performance. |
Tab. 1: For medical device software, and thus also AI-based medical device software, three aspects of clinical evidence must be substantiated.
1) For CTs, brightness values correspond to Hounsfield Units (HU), sometimes referred to as "X-ray density".
b) Detection methods
The MDCG 2020-1 guideline suggests different methods to demonstrate the above aspects.
Aspect to be proven | Methods (Examples) |
clinical association/scientific validity | systematic literature search in medical guidelines and scientific literature (for example, it is described which "brightness values" cancer tissue has) clinical investigations or clinical performance studies for IVD/MD software |
technical performance/analytical performance | system-level software testing to demonstrate the following performance parameters: analytical sensitivity, analytical specificity, trueness (bias), precision (repeatability and reproducibility), accuracy (as an output of trueness and precision), limits of detection and quantification, measurement range, linearity, cut-off (These tests are performed in addition to or based on the unit and integration tests according to IEC 62304, the proof of IT security and usability; cf. the criteria of ISO 25010.) |
clinical performance | retrospective analyses of existing data in sufficient quality and quantity clinical investigations (or clinical performance studies for IVD/MD software) clinical literature (assumes equivalence - including clinical equivalence) can be used as a baseline or supplemental, as appropriate |
Tab. 2: For each aspect, it is important to use the right methods
Note
Validation of AI software against its intended purpose includes usability testing (summative evaluation) to demonstrate usability for purpose in addition to clinical performance evidence.
Further information
The GHTF document Software as a Medical Device: Clinical Evaluation lists further methods and explains the aspects to be demonstrated.
5. Collecting clinical data
Demonstrating clinical performance is the biggest challenge for manufacturers of AI-based medical devices. However, this does not mean that prospective clinical investigations are always necessary.
a) Retrospective analyses
In a retrospective analysis, manufacturers use existing data.
In the case of cancer detection software, the manufacturer could use existing CT scans, have the software evaluate them, and compare them to existing cancer diagnoses. However, this only works if the following conditions are met:
- The images were obtained under comparable technical conditions as intended by normal use. This includes the acquisition equipment (CTs), acquisition parameters (e.g., radiation intensity, angle, patient positioning, with/without contrast agent), and image parameters (size and resolution of images, coding of Hounsfield values)
- The patient population with which the images were recorded is comparable to that intended in the intended purpose. This includes anatomic and morphologic characteristics as well as different locations, sizes, and types of tumors and possible effusions.
- The number of cases is sufficient, and the quality of the data is scientifically valid.
- Informed consent has been obtained to use the data for clinical study purposes.
- Comparability of users and the use environment would also need to be discussed. However, usability tests are easier to replicate than clinical investigations.
b) Clinical investigations in artificial intelligence
If these requirements are not met or not fully met, manufacturers must generate the clinical data, typically through a prospective clinical investigation (or clinical performance study for IVD/MD software).
This would be necessary, for example, if the software is also to be used for patients with cancer for whom sufficient clinical data are not yet available. In this specific case, the clinical investigation would at least have to demonstrate the correct segmentation of the cancer tissue for this patient population, which has not been reviewed so far.
6. Conclusion
Attempts to demonstrate the equivalence of AI models and to prove the clinical performance of the software based on the literature usually fail. Many manufacturers even explicitly claim "better" models.
However, this does not mean that a prospective clinical investigation is necessarily required for AI-based medical devices. It is often possible to avoid these trials in whole or in part by retrospective analyses of already available data of sufficient quality and quantity.
Tip
A smartly chosen clinical strategy or performance evaluation strategy and a focused intended use will help to bring the device to market faster. During the subsequent use of the device, clinical data is generated, which you can evaluate, e.g., as PMCF or PMPF studies, and use for an extension of the intended purpose.
Contact us to work with the Johner Institute's clinical experts on the best clinical strategy or performance evaluation strategy for your device. This will save you time and money (e.g., for clinical investigations in artificial intelligence) and enable you to bring your device to market quickly and compliantly.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.