Regulatory requirements for medical devices with machine learning
The incorporation of AI in medical devices has made great strides, for example, in the diagnosis of disease. Manufacturers of devices with machine learning face the challenge of having to demonstrate compliance of their devices with the regulations.
Even if you know the law - what are the standards and best practices to consider in order to provide the evidence and speak to authorities and notified bodies on the same level?
This article provides an overview of the most important regulations and best practices that you should consider. You can save yourself the trouble of researching and reading hundreds of pages and be perfectly prepared for the next audit.
1. Legal requirements for the use of machine learning in medical devices
a) MDR and IVDR
Currently, there are no laws or harmonized standards that specifically regulate the use of machine learning in medical devices. However, it appears that these devices must comply with existing regulatory requirements such as MDR and IVDR, e.g.:
- Manufacturers must demonstrate the benefits and performance of the medical devices. For example, devices used for diagnosis require proof of diagnostic sensitivity and specificity.
- The MDR requires manufacturers to ensure the safety of devices. This includes ensuring that software is designed to ensure repeatability, reliability, and performance (see MDR Annex I, 17.1 or IVDR Annex I, 16.1).
- Manufacturers must formulate a precise intended use (MDR/IVDR Annex II). They must validate their devices against the intended use and stakeholder requirements and verify them against the specifications (including MDR Annex I, 17.2 or IVDR Annex I, 16.2). Manufacturers are also required to describe the methods they use to provide this evidence.
- If the clinical evaluation is based on a comparative product, there must be technical equivalence, which explicitly includes the evaluation of the software algorithms (MDR Annex XIV, Part A, paragraph 3). This is even more difficult in the case of performance evaluation of in vitro diagnostic devices (IVDs). Only in well-justified cases can a clinical performance study be waived (IVDR Annex XIII, Part A, paragraph 1.2.3).
- The development of the software that will become part of the device must take into account the "principles of development life cycle, risk management, including information security, verification and validation" (MDR Annex I, 17.2 or IVDR Annex I, 16.2).
b) EU AI Regulation (AI Act)
i) The scope
On its webpage New rules for Artificial Intelligence - Questions and Answers the EU announces its intention to regulate AI in all sectors on a risk-based basis and to monitor compliance with the regulations. You can find the Proposal for a Regulation laying down harmonized rules on artificial intelligence (Artificial Intelligence Act) here.
The EU sees the opportunities of using AI but also its risks. It explicitly mentions healthcare and wants to prevent fragmentation of the single market through EU-wide regulation.
Regulation should affect both manufacturers and users. The regulations to be implemented depend on whether the AI system is classified as a high-risk AI system or not. Devices covered by the MDR or IVDR that require the involvement of a notified body are considered high-risk.
One focus of regulation will be on remote biometric identification.
The planned regulation also provides for monitoring and auditing; in addition, it extends to imported devices. The EU plans for this regulation to interact with the new Machinery Regulation, which would replace the current Machinery Directive.
A special committee is to be established: "The European Committee on Artificial Intelligence is to be composed of high-level representatives of the competent national supervisory authorities, the European Data Protection Supervisor, and the Commission".
Further information
For the latest news on AI regulation, visit the EU website. It has also launched the European AI Alliance.
ii) The criticism
As always, the EU emphasizes that the location should be promoted, and SMEs should not be unduly burdened. These statements can also be found in the MDR. But none of the regulations seem to meet this requirement.
Content of the AI Regulation | Unwanted consequences |
Artificial Intelligence procedures include not only machine learning but also: "- Logic and knowledge-based approaches, including knowledge representation, inductive (logic) programming, knowledge bases, inference and deduction engines, (symbolic) reasoning and expert systems; - Statistical approaches, Bayesian estimation, search, and optimization methods." | This ready scope may result in many medical devices containing software falling within the scope of the AI Regulation. Accordingly, any decision tree cast in software would be an AI system. |
The AI Regulation explicitly addresses medical devices and IVDs. | There is a duplication of requirements. MDR and IVDR already require cybersecurity, risk management, post-market surveillance, a notification system, technical documentation, a QM system, etc. Manufacturers will soon have to demonstrate compliance with two regulations! |
The regulation applies regardless of what the AI is used for in the medical device. | Even an AI that is intended to realize the lower-wear operation of an engine would fall under EU regulation. As a consequence, manufacturers will think twice before making use of AI procedures. This can have a negative impact on innovation but also on the safety and performance of devices. This is because, as a rule, manufacturers use AI to improve the safety, performance, and effectiveness of devices. Otherwise, they would not be allowed to use AI at all. |
The regulation requires: "High-risk AI systems shall be designed and developed in such a way that they can be effectively supervised by natural persons during the period of use of the AI system, including with appropriate tools of a human-machine interface." | This requirement rules out the use of AI in situations in which humans can no longer react quickly enough. Yet, it is precisely in these situations that the use of AI could be particularly helpful. If we have to place a person next to each device to "effectively supervise" the use of AI, this will mean the end of most AI-based products. |
The regulation defines the crucial term "safety component" by using the undefined term of a safety function: A "safety component of a product or system" is a "component of a device or system that performs a safety function for that device or system, or whose failure or malfunction endangers the health and safety of persons or property;" The AI Regulation also does not define other terms in accordance with the MDR, e.g., "post-market monitoring" or "serious incident." | There will be disputes about what constitutes a safety function. For example, it could be a function that puts the safety of patients at risk if it behaves out of specification. But it could also mean a function that implements a risk-minimizing measure.
Definitions that are not aligned increase the effort required by manufacturers to understand and align the various concepts and associated requirements. |
A device counts as a high-risk AI system if both of the following conditions are met: "(a) the AI system is intended to be used as a safety component of a device covered by Union harmonization legislation listed in Annex II or is itself such a device; (b) the device of which the AI system is the safety component, or the AI system itself as a device, shall be subject to a third-party conformity assessment with regard to placing on the market or putting into service that device in accordance with the Union harmonization legislation listed in Annex II." | Medical devices are covered by the regulations listed in Annex II, as they mention the MDR and IVDR. Medical devices of class IIa and higher must undergo a conformity assessment procedure. Does this make them high-risk devices?
The unfortunate Rule 11 classifies software - regardless of risk - into Class IIa or higher in the vast majority of cases. This means that medical devices are subject to the extensive requirements for high-risk products. The negative effects of Rule 11 are reinforced by the AI Regulation. |
In Article 10, the AI Regulation requires | Real-world data is rarely error-free and complete. It also remains unclear what is meant by "complete." Do all data sets have to be present (whatever that means) or all data of a data set? |
Article 64 of the AI Regulation requires manufacturers to provide authorities with full remote access to training, validation, and testing data, even through an API. | Making confidential patient data accessible via remote access is in dispute with the legal requirement of data protection by design. Health data belongs to the personal data category that requires special protection. Developing and providing an external API to the training data means an additional effort for the manufacturers. That authorities with this access can download, analyze, and evaluate the data or AI with reasonable effort and time is unrealistic. For other, often even more critical data and information on product design and production (e.g., source code or CAD drawings), no one would seriously require manufacturers to provide remote access to authorities. |
Tab. 1: Demands of the AI Act
The Johner Institute had submitted a statement with these concerns to the EU.
iii) News on the AI Act
A new draft of the AI Act has been available since October 2022. This resolves many inconsistencies and removes unclear requirements. But criticisms that we also reported to the EU remain:
- Overly broad scope of the Act (due to the definition of "artificial intelligence systems").
- Duplications and inconsistencies with the requirements of MDR and IVDR.
The fact that now, of all things, public institutions such as law enforcement agencies are to be exempted from the obligation to register AI-based devices could generate suspicion.
However, it is to be welcomed that AI systems that are used exclusively for research are exempt from regulation.
The EU was able to reach a compromise by the end of 2023. This compromise is now available in an almost 900-page document that is still difficult to read.
The AI Act was published in March 2024.
Download
You can find the adopted version of the AI Act on the EU website, although it still needs to be edited.
c) (Harmonized) standards without specific reference to machine learning
The MDR and IVDR allow proof of conformity to be provided with the aid of harmonized standards and common specifications. In the context of medical devices that use machine learning procedures, manufacturers should pay particular attention to the following standards:
- ISO 13485:2016
- IEC 62304
- IEC 62366-1
- ISO 14971
- IEC 82304
These standards contain specific requirements that are also relevant for medical devices with machine learning, e.g.:
- The development of software for data collection and reprocessing, labeling, and model training and inspection must be validated (Computerized Systems Validation (CSV) according to ISO 13485:2016 4.16).
- Manufacturers shall determine and ensure the competence of those involved in the development prior to development (ISO 13485:2016 7.3.2 f).
- IEC 62366-1 requires that manufacturers accurately characterize the intended users and the intended use environment, as well as the patients, including indications and contraindications.
- Manufacturers who use software libraries (which should almost always be the case for machine learning software) must specify and validate these libraries as SOUP/OTS (IEC 62304).
Further information
Please consider the article on validation of ML libraries.
2. Legal requirements for the use of machine learning in medical devices in the USA
a) Unspecific requirements
The FDA has similar requirements, especially in 21 CFR part 820 (including part 820.30 with the design controls). There are numerous guidance documents, including those on software validation, the use of off-the-shelf software (OTSS), and cybersecurity. These are required reading for companies that want to sell medical devices in the US that are or contain software.
b) Specific requirements Part 1 (Framework 2019 - meanwhile outdated)
In April 2019, the FDA published a draft Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD).
In it it talks about the in-house requirements for continuous learning systems. The FDA notes that the medical devices approved to date that are based on AI procedures work with "locked algorithms."
For the changes to the algorithms, the authority would like to explain when it
- does not expect a new submission but only documentation of the changes by the manufacturer,
- would at least like to carry out a review of the changes and validation before the manufacturer is allowed to place the modified device on the market,
- insists on a (completely) new submission or approval.
Existing approaches
The new framework is based on existing approaches:
- IMDRF risk categories for Software as Medical Device (SaMD)
- the FDA's benefit-risk framework
- FDA considerations on when software changes require reauthorization (software changes)
- approval procedures, including the FDA's Pre-Cert program, de novo procedures, etc.
- FDA guidance on the clinical evaluation of software
What the objectives of changes to an algorithm can be
According to FDA rules, an algorithm that is self-learning or continues to learn during use must be subject to review and approval. This seems too rigid even for the FDA. Therefore, it is examining the objectives of changing the algorithm and distinguishing:
- Improvement of clinical and analytical performance: this improvement could be achieved by training with more data sets.
- Changing the "input data" that the algorithm processes: this could be additional laboratory data or data from another CT manufacturer.
- Changing the intended use: as an example, the FDA states that the algorithm initially only calculates a "confidence score" to support the diagnosis and later calculates the diagnosis directly. A change in the intended patient population also counts as a change of intended use.
Depending on these objectives, the authority would like to decide on the need for new submissions.
Pillars of a best practice approach
The FDA names four pillars that manufacturers should use to ensure the safety and benefit of their devices throughout the product life cycle, even in the event of changes:
- Quality Management System and Good Machine Learning Practices (GMLP)
The FDA expects clinical validity to be ensured. (You can find out what this is in this article.) However, this requirement is not specific to AI algorithms. The FDA does not name specific GMLPs. It only mentions an appropriate separation of training, "tuning," and test data, as well as appropriate transparency about the output and the algorithms. - Planning and initial assessment of safety and performance
In comparison to "normal" approvals, the FDA expects, among other things, "SaMD Pre-Specifications" (SPS), in which manufacturers explain which categories of changes (see above) they anticipate. In addition, they should make changes in accordance with an "Algorithm Change Protocol" (ACP). This is not a protocol but a procedure. The subject of this procedure is shown in Fig. 1. - Approach to evaluate changes after initial release
If manufacturers did not submit an SPS and ACP at the time of initial approval, they must resubmit future changes to the authority. Otherwise, the authority decides whether it expects a resubmission, whether it "only" performs a "focused review," or whether it expects the manufacturer to document the changes. The decision depends on whether the manufacturer follows the "approved" SPS and ACP and/or whether the intended use changes (see Fig. 2). - Transparency and monitoring of market performance
The FDA expects regular reports on the monitoring of the performance of devices in the market according to the SPS and ACP. Users would also have to be informed about which changes the manufacturer has made and how they affect performance, for example.
By transparency, the FDA, therefore, does not mean explaining how the algorithms work "under the hood," for example, but openness about what the manufacturer has changed, with what purpose, and what effects.

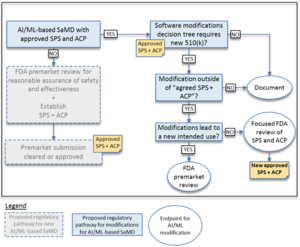
Example of the circumstances in which the authority does (not) have to be involved in changes
The FDA gives examples of when a manufacturer may change the algorithm of software without asking the authority for approval. The first of these examples is software that predicts imminent patient instability in an intensive care unit based on monitor data (e.g., blood pressure, ECG, pulse oximeter).
The manufacturer plans to change the algorithm, e.g., to minimize false alarms. If he already provided for this in the SCS and had it approved by the authority together with the ACP, he may make these changes without renewed "approval."
However, if he claims that the algorithm provides 15 minutes of warning of physiologic instability (he now additionally specifies a time duration), that would be an expansion of the intended use. This change would require FDA approval.
Summary
The FDA is debating how to deal with continuous learning systems. It has not even answered the question of what best practices are for evaluating and approving a "frozen" algorithm based on AI procedures.
There is still no guidance document that defines what the FDA calls "Good Machine Learning Practices." The Johner Institute is therefore developing such a guideline together with a notified body.
The FDA's concept of waiving the need for a new submission based on pre-approved procedures for changes to algorithms has its charm. You will look in vain for such concreteness on the part of European legislators and authorities.
Specific requirements Part 2 (Framework 2023)
The FDA's "Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD)" is also mandatory reading. In April 2023, the FDA converted this into a guidance document entitled "Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence/Machine Learning (AI/ML)-Enabled Device Software Functions."
Another FDA guidance document on radiological imaging does not directly address AI-based medical devices; however, it is helpful. First, many AI/ML-based medical devices work with radiological imaging data, and second, the document identifies sources of error that are particularly relevant to ML-based devices as well:
- Patient characteristics
Demographic and physiological characteristics, motion artifacts, implants, spatially heterogeneous distribution of tissue, calcifications, etc. - Imaging characteristics
Positioning, specific characteristics of medical devices, acquisition parameters (e.g., sequences for MRIs or X-ray doses for CTs), image reconstruction algorithms, external sources of interference, etc. - Image processing
Filtering, different software versions, manual selection and segmentation of areas, fitting of curves, etc.
The FDA, Health Canada, and the UK Medicines and Healthcare Products Regulatory Agency (MHRA) have collaborated to publish "Good Machine Learning Practice for Medical Device Development: Guiding Principles". The document contains ten guiding principles that should be followed when using machine learning in medical devices. Due to its brevity of only two pages, the document does not go into detail, but it does get to the heart of the most important principles.
3. Legal requirements for the use of machine learning for medical devices in other countries
a) China: NMPA
The Chinese NMPA has released the draft "Technical Guiding Principles of Real-World Data for Clinical Evaluation of Medical Devices" for comment.
However, this document is currently only available in Chinese. We have had the table of contents translated automatically. The document addresses:
- Requirements analysis
- Data collection and reprocessing
- Design of the model
- Verification and validation (including clinical validation)
- Post-Market Surveillance
Download: China-NMPA-AI-Medical-Device
The authority is expanding its staff and has established an AI Medical Device Standardization Unit. This unit is responsible for standardizing terminologies, technologies, and processes for development and quality assurance.
b) Japan
The Japanese Ministry of Health, Labor, and Welfare is also working on AI standards. Unfortunately, the authority publishes the progress of these efforts only in Japanese. (Translation programs are helpful, however.) Concrete output is still pending.
4. Standards and best practices relevant for machine learning
a) „Artificial Intelligence in Healthcare“ by COICR
From April 2019 is COICR's Artificial Intelligence in Healthcare document. It does not provide specific new requirements but refers to existing ones and recommends the development of standards.
Conclusion: Not very helpful
b) IEC/TR 60601-4-1
Technical Report IEC/TR 60601-4-1 provides specifications for "medical electrical equipment and medical electrical systems with a degree of autonomy." However, these specifications are not specific to medical devices that use machine learning procedures.
Conclusion: Conditionally helpful
c) „Good Practices“ by Xavier University
“Perspectives and Best Practices for AI and Continuously Learning Systems in Healthcare" was published by Xavier University.
As the title makes clear, it (also) deals with continuous learning systems. Nevertheless, many of the best practices mentioned can also be applied to non-continuously learning systems:
- Initially define the requirements for performance
- Gather information and understand how the system learns over time
- Follow professional software development process, including verification and validation
- Perform systematic quality control on new data with which the system is to (continue to) learn
- Set limits within which the algorithm is allowed to change over time
- Determine what changes to the algorithm are allowed to trigger
- Design the system to report its own performance in a timely manner and to report the output to the user at regular intervals
- Provide users with the ability to refuse to update an algorithm and/or revert to a previous version
- Inform users when learning has caused a significant change in behavior, and clearly describe that change
- Make it traceable how an algorithm has evolved and how it came to a decision
This traceability/interpretability, in particular, is a challenge for many manufacturers.
Further information
The video training at the Medical Device University introduces important procedures like LRP LIME, visualization of neural layer activation, or counterfactuals.
The document also discusses exciting issues, such as whether patients need to be informed when an algorithm has evolved and may subsequently arrive at a better or even different diagnosis.
Guidelines from this document will be incorporated into the Johner Institute's AI Guide.
Conclusion: Helpful especially for continuously learning systems
d) „Building Explainability and Trust for AI in Healthcare“ by Xavier University
This document from Xavier University, to which the Johner Institute also contributed, addresses best practices in the area of explainability. It provides useful guidance on what information needs to be provided, e.g., to technical stakeholders, in order to meet the requirements for explainability.
Conclusion: At least partially helpful
e) „Machine Learning AI in Medical Devices“ by BSI and AAMI
The title of this BSI/AAMI document sounds promising. Ultimately, however, it is just a position paper that you can download free of charge from the AAMI Store. The position paper calls for the development of more standards in which BSI and AAMI are participating. One is the standard BS/AAMI 34971:2023-06-30, which introduces subchapter 4. r).
f) DIN SPEC 92001-1:2019-04
The standard DIN SPEC 92001 "Artificial Intelligence - Life Cycle Processes and Quality Requirements - Part 1: Quality Meta Model" is even available free of charge.
It presents a meta-model but does not specify any concrete requirements for the development of AI/ML systems. The document is completely non-specific and not targeted to any particular industry.
Conclusion: Not very helpful
g) DIN SPEC 9200-2 (still in development)
“Part 2: Robustness" is not yet available. In contrast to the first part, it contains concrete requirements. These are aimed primarily at risk management. However, they are not specific to medical devices.
Conclusion: To be observed, promising
h) ISO/IEC CD TR 29119-11
The standard ISO/IEC TR 29119-11 "Software and systems engineering - Software testing - Part 11: Testing of AI-based systems" is still under development.
We have read and evaluated this standard for you.
Conclusion: Ignore
i) Curriculum of the Korean Software Testing Qualification Board
The "International Software Testing Qualification Board" (ISTQB) provides a syllabus for testing AI systems with the title "Certified Tester AI Testing (CT-AI) Syllabus" for download.
Chapters 1 through 3 explain terms and concepts. Chapter 4 explicitly addresses data management. Chapter 5 defines performance metrics. Starting in Chapter 7, the syllabus provides guidance on testing AI systems.
In addition, Chapter 9 of the document provides guidelines for black-box testing of AI models, such as combinatorial testing and "metamorphic testing." Tips for neural network testing, such as "Neuron Coverage," and tools, such as DeepXplore, are also worth mentioning.
Conclusion: Recommended
j) ANSI/CTA standards
ANSI, together with the CSA (Consumer Technology Association), has published several standards:
- Definitions and Characteristics of Artificial Intelligence (ANSI/CTA-2089)
- Definitions/Characteristics of Artificial Intelligence in Health Care (ANSI/CTA-2089.1)
The standards provide - as the title suggests - definitions. Nothing more and nothing less.
The CSA is currently working on further and concrete standards, including one on "trustworthiness."
Conclusion: Only helpful as a collection of definitions
k) IEEE standards
A whole family of standards is under development at IEEE:
- P7001 – Transparency of Autonomous Systems
- P7002 – Data Privacy Process
- P7003 – Algorithmic Bias Considerations
- P7009 – Standard for Fail-Safe Design of Autonomous and Semi-Autonomous Systems
- P7010 – Wellbeing Metrics Standard for Ethical Artificial Intelligence and Autonomous Systems
- P7011 – Standard for the Process of Identifying and Rating the Trustworthiness of News Sources
- P7014 – Standard for Ethical considerations in Emulated Empathy in Autonomous and Intelligent Systems
- 1 – Standard for Human Augmentation: Taxonomy and Definitions
- 2 – Standard for Human Augmentation: Privacy and Security
- 3 – Standard for Human Augmentation: Identity
- 4 – Standard for Human Augmentation: Methodologies and Processes for Ethical Considerations
- P2801 – Recommended Practice for the Quality Management of Datasets for Medical Artificial Intelligence
- P2802 – Standard for the Performance and Safety Evaluation of Artificial Intelligence Based Medical Device: Terminology
- P2817 – Guide for Verification of Autonomous Systems
- 1.3 – Standard for the Deep Learning-Based Assessment of Visual Experience Based on Human Factors
- 1 – Guide for Architectural Framework and Application of Federated Machine Learning
Conclusion: Still too early, keep watching
l) ISO standards under development
Several working groups at ISO are also working on AI/ML-specific standards:
- ISO 20546 – Big Data – Overview and Vocabulary
- ISO 20547-1 – Big Data reference architecture – Part 1: Framework and application process
- ISO 20547-2 – Big Data reference architecture – Part 2: Use cases and derived requirements
- ISO 20547-3 – Big Data reference architecture – Part 3: Reference architecture
- ISO 20547-5 – Big Data reference architecture – Part 5: Standards roadmap
- ISO 22989 – AI Concepts and Terminology
- ISO 24027 – Bias in AI systems and AI aided decision making
- ISO 24029-1 – Assessment of the robustness of neural networks – Part 1 Overview
- ISO 24029-2 – Formal methods methodology
- ISO 24030 – Use cases and application
- ISO 24368 – Overview of ethical and societal concerns
- ISO 24372 – Overview of computations approaches for AI systems
- ISO 24668 – Process management framework for Big data analytics
- ISO 38507 – Goveranance implications of the use of AI by organizations.
The first standards have already been completed (such as the one presented below).
Conclusion: Still too early, keep watching
m) ISO 24028 – Overview of trustworthiness in AI
ISO/IEC TR 24048 is entitled "Information Technology - Artificial Intelligence (AI) - Overview of trustworthiness in artificial intelligence". It is unspecific to a particular domain, but gives examples, including healthcare.
The standard summarizes important hazards and threats as well as common risk mitigation measures (see Fig. 1).
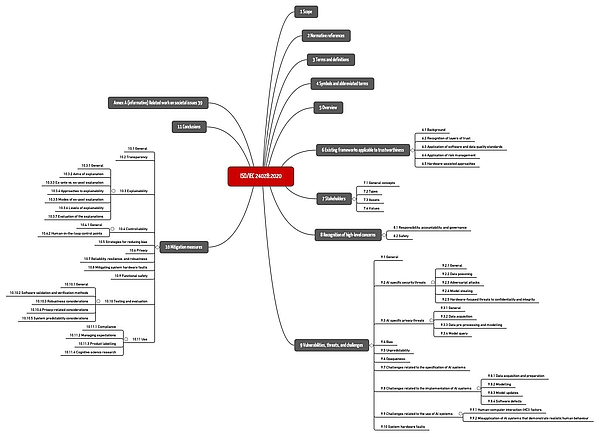
Download: ISO-IEC-24028-2020: Mindmap of the chapter structure
However, the standard remains at a general level, does not provide any concrete recommendations, and does not set any specific requirements. It is useful as an overview and introduction as well as a reference to further sources.
Conclusion: Conditionally recommendable
n) ISO 23053 – Framework for AI using ML
ISO 23053 is a guideline for a development process of ML models. It contains no specific requirements but represents the state of the art.
Conclusion: Conditionally recommendable
o) AI4H guidance by WHO/ITU
Specific to healthcare, the WHO and ITU (International Telecommunication Union) are developing a framework for the use of AI in healthcare, particularly for diagnosis, triage, and treatment support.
This AI4H initiative includes several Topic Groups from different medical faculties as well as Working Groups addressing cross-cutting issues. The Johner Institute is an active member of the Working Group on Regulatory Requirements.
This Working Group is developing a guidance document that will build on and potentially supersede the previous Johner Institute guidance document. Coordination of these outputs with IMDRF is planned.
To learn more about this initiative, contact ITU or the Johner Institute.
Conclusion: Highly recommended in the future
p) Guide of the notified bodies
The notified bodies have developed a guide to Artificial Intelligence based on the Johner Institute's guide. Since this is published and used by the notified bodies, it is a must-read, at least for German manufacturers.
Conclusion: Highly recommended
q) Key terms and definitions by the IMDRF
The International Medical Device Regulators Forum (IMDRF) proposed a document with key terms and definitions for "Machine Learning-enabled Medical Devices - A subset of Artificial Intelligence-enabled Medical Devices" on September 16, 2021. The consultation period ends on November 29, 2021.
Conclusion: Could become helpful by standardizing terms
r) BS/AMMI 34971:2023
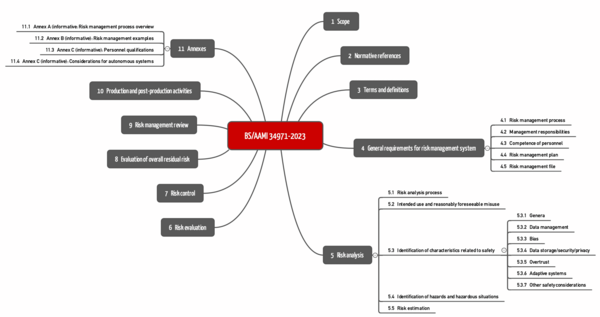
BS/AAMI 34971 has been available since May 2023. It is entitled "Application of ISO 14971 to machine learning in artificial intelligence" and can be obtained, for example, from Beuth for more than 250 EUR.
The standard strictly follows the structure of ISO 14971 (see Fig.). The chapters on the third level are specific. These can be found below in chapter 5.3 ("Identification of characteristics related to safety").
What pleases
The standard is strictly based on ISO 14971. This facilitates (theoretically) the assignment.
The examples given in the standard are very comprehensive. They can thus serve as a valuable checklist for identifying and eliminating possible causes of hazards.
The standard also lists helpful measures for risk control and, for example, specific requirements for the competence of personnel in the appendix.
What we would have liked to see differently
The authors cannot be held responsible for the price. Nevertheless, we would have liked BS/AAMI 34971 not to be more expensive than the standard to which it refers.
It seems as if the authors of BS/AAMI 34971 understand some concepts differently than the authors of ISO 14971.
For example, the chapter "Identification of characteristics related to safety" includes dozens of examples, but they are not safety characteristics but causes of hazards (e.g., the bias in the data). This is very unfortunate because
- this contributes to confusion, which can negatively impact the efficiency and effectiveness of audits and reviews and make it difficult to map into algorithms,
- this seems to repeat exactly the mistake that manufacturers of ML-based devices keep making: They fail to derive the essential performance or safety-related characteristics from the intended use first, and only then derive the requirements for the ML models.
Elsewhere, it is claimed that Table B.2. of BS/AAMI 34971 corresponds to Table C2 of ISO 14971. The first is headed "Events and circumstances" (?!?), the second "Hazards". Why do the authors of BS/AAMI 34971 introduce new terms and concepts that they do not define but seem to equate with defined terms?
Further, it is unfortunate that explanations and requirements are not precisely separated. It seems as if the concepts of "Ground Truth" and "Gold Standard" are not neatly distinguished. The term "ML validation test" can only be understood from the context. For ML experts, "validation" and "test" are two different activities.
Statisticians will certainly form their own opinion about risk-minimizing measures such as "denoising of data."
Conclusion: If 250 EUR is no obstacle, buy the standard and use it as a checklist and source of inspiration
There is also BS ISO/IEC 23894:2023 "Information technology. Artificial intelligence. Guidance on risk management". This is not a duplication of BS 39471, as this standard is not specific to medical devices.
s) ISO/NP TS 23918
ISO/NP TS 23918, "Medical devices - Guidance on the application of ISO 14971 - Part 2: Machine learning in artificial intelligence," is still under development. Its scope of application appears to be very similar to that of AAMI/BS 34971, which also deals with applying ISO 14971 to AI-based medical devices.
t) BS 30440:203
BSI also issued the standard BS 30440:2023 entitled "Validation framework for the use of artificial intelligence (AI) within healthcare. Specification".
It is interesting to note that this standard sees not only manufacturers but also operators, health insurers, and users as readers.
u) ISO/IEC 23894
ISO/IEC 23894 was published in February 2023 and is entitled "Artificial intelligence - Guidance on risk management." The standard cannot be used without ISO 31000:2018 (which is the standard on "general risk management," i.e., not medical device-specific). It is seen more as a delta that complements the AI-specific aspects. Real assistance for companies using AI in their medical devices cannot be discovered immediately.
Conclusion: Do not buy
v) ISO/IEC 42001:2023
The standard ISO/IEC 42001:2023 is entitled "Information technology - Artificial intelligence - Management system".
This makes the scope clear: it deals with the requirements for a management system. Its scope includes the use of AI within the organization as well as AI-based devices of this organization. However, the standard is not specific to medical devices (manufacturers).
Overall, the standard is very "high-level" and too unspecific, especially for developing AI-based devices. This is not surprising because the standard is a process standard, not a product one.
Many of the requirements are already met by organizations that comply with the requirements of ISO 13485 and ISO 14971.
The standard will become more important as AI becomes part of companies' everyday lives. The "AI system life cycle" approach is certainly the right one.
5. Questions you should be prepared for in the audit
a) General
Notified bodies and authorities have not yet agreed on a uniform approach and common requirements for medical devices with machine learning.
As a result, manufacturers regularly struggle to prove that the requirements placed on the device are met, for example, in terms of accuracy, correctness, and robustness.
Dr. Rich Caruana, one of Microsoft's leaders in Artificial Intelligence, even advised against using a neural network he developed himself to suggest the appropriate therapy for patients with pneumonia:
„I said no. I said we don’t understand what it does inside. I said I was afraid.”
Dr. Rich Caruana, Microsoft
That there are machines that a user does not understand is not new. You can use a PCR without understanding it; there are definitely people who know how this device works and its inner workings. However, with Artificial Intelligence, that is no longer a given.
b) Key questions
Questions auditors should ask manufacturers of machine learning devices include:
Key question | Background |
Why do you think your device is state of the art? | Classic introductory question. Here you should address technical and medical aspects. |
How do you come to believe that your training data has no bias? | Otherwise, the outputs would be incorrect or correct only under certain conditions. |
How did you avoid overfitting your model? | Otherwise, the algorithm would correctly predict only the data it was trained with. |
What leads you to believe that the outputs are not just randomly correct? | For example, it could be that an algorithm correctly decides that a house can be recognized in an image. However, the algorithm did not recognize a house but the sky. Another example is shown in Fig. 3 |
What conditions must data meet in order for your system to classify them correctly or predict the outputs correctly? Which boundary conditions must be met? | Because the model has been trained with a specific set of data, it can only make correct predictions for data that come from the same population. |
Wouldn't you have gotten a better output with a different model or with different hyperparameters? | Manufacturers must minimize risks as far as possible. This includes risks from incorrect predictions of suboptimal models. |
Why do you assume that you have used enough training data? | Collecting, reprocessing, and "labeling" training data is costly. The larger the amount of data with which a model is trained, the more powerful it can be. |
Which standard did you use when labeling the training data? Why do you consider the chosen standard as gold standard? | Especially when the machine begins to outperform humans, it becomes difficult to determine whether a doctor, a group of "normal" doctors, or the world's best experts in a specialty are the reference. |
How can you ensure reproducibility as your system continues to learn? | Especially in Continuously Learning Systems (CLS) it has to be ensured that the performance does at least not decrease due to further training. |
Have you validated systems that you use to collect, prepare, and analyze data, as well as train and validate your models? | An essential part of the work consists of collecting and reprocessing the training data and training the model with it. The software required for this is not part of the medical device. However, it is subject to the requirements for Computerized Systems Validation. |
Tab. 2: Potential issues in medical device review with associated declaration
The above issues are typically also discussed in the context of risk management according to ISO 14971 and clinical evaluation according to MEDDEV 2.7.1 Revision 4 (or performance evaluation of IVDs).
Further information
For guidance on how manufacturers can use Machine Learning to meet these regulatory requirements for medical devices, see the article on Artificial Intelligence in Medicine.
6. Typical mistakes made by AI startups
Many startups that benefit from Artificial Intelligence (AI) procedures, especially Machine Learning, begin product development with the data. In the process, they often make the same mistakes:
Mistake | Consequences |
The software and the processes for collecting and reprocessing the training data have not been validated. Regulatory requirements are known in rudimentary form at best. | In the worst case, the data and models cannot be used. This throws the whole development back to the beginning. |
The manufacturers do not derive the declared performance of the devices from the intended use and the state of the art but from the performance of the models. | The devices fail clinical evaluation. |
People whose real passion is data science or medicine try their hand at enterprise development. | The devices never make it to market or don't meet the real need. |
The business model remains too vague for too long. | Investors hold back or/and the company dries up financially and fails. |
Tab. 3: Typical mistakes made by AI startups and their consequences
Startups can contact us. In a few hours, we can help avoid these fatal mistakes.
7. Summary and conclusion
a) Regulatory requirements
The regulatory requirements are clear. However, it remains unclear to manufacturers, and in some cases also to authorities and notified bodies, how these are to be interpreted and implemented in concrete terms for medical devices that use machine learning procedures.
b) Too many and only conditionally helpful best practice guides
As a result, many institutions feel called upon to help with "best practices." Unfortunately, many of these documents are of limited help:
- Reiterate textbook knowledge about artificial intelligence in general and machine learning in particular.
- The guidance documents get bogged down in self-evident facts and banalities. Anyone who didn't know before reading these documents that machine learning can lead to misclassification and bias, putting patients at risk or at a disadvantage, should not be developing medical devices.
- Many of these documents are limited to listing issues specific to machine learning that manufacturers must address. Best practices on how to minimize these problems are lacking.
- When there are recommendations, they are usually not very specific. They do not provide sufficient guidance for action.
- It is likely to be difficult for manufacturers and authorities to extract truly testable requirements from textual clutter.
Unfortunately, there seems to be no improvement in sight; on the contrary, more and more guidelines are being developed. For example, the OECD recommends the development of AI/ML-specific standards and is currently developing one itself. The same is true for the IEEE, DIN, and many other organizations.
Conclusion:
- There are too many standards to keep track of. And there are continuously more.
- The standards overlap strongly and are predominantly of limited benefit. They do not contain (binary decidable) test criteria.
- They come (too) late.
c) Quality instead of quantity
In machine learning best practices and standards, medical device manufacturers need more quality, not quantity.
Best practices and standards should guide action and set verifiable requirements. The fact that WHO is taking up the Johner Institute's guidance is cause for cautious optimism.
It would be desirable if the notified bodies, the authorities, and, where appropriate, the MDCG were more actively involved in the (further) development of these standards. This should be done in a transparent manner. We have recently experienced several times what modest outputs are achieved by working in backrooms without (external) quality assurance.
With a collaborative approach, it would be possible to reach a common understanding of how medical devices that use machine learning should be developed and tested. There would only be winners.
Notified bodies and authorities are cordially invited to participate in further developing the guidelines. An e-mail to the Johner Institute is sufficient.
Manufacturers who would like support in the development and approval of ML-based devices (e.g., in the review of technical documentation or in the validation of ML libraries) are welcome to contact us via e-mail or the contact form.
Change History
- 2024-03-26: Link to the passed AI Act added
- 2024-01-18: Added chapter 4.v) that evaluates ISO 42001 and note on the compromise proposal on the AI Act added
- 2023-11-10: Chapter structure revised. FDA requirements added. New BS standards mentioned. New chapter 4.s) with ISO/NP TS 23918 added
- 2023-10-09: Added chapter 2.n) that evaluates ISO 23053
- 2023-09-07: Added chapter 2.s) that evaluates AAMI 34971:2023-05-30
- 2023-07-03: Added link to BS/AAMI 34971:2023-05-30
- 2023-06-27: Added section 1(b)(i): AI act applies to MP/IVD of higher risk classes
- 2023-06-23: FDA Guidance Document from April 2023 added
- 2023-06-08: Chapter 4 added
- 2023-02-14: Section 2.r) added
- 2022-11-17: Link to new draft of AI Act added
- 2022-10-21: Added link to EU updates on AI Act
- 2022-06-27: Added in chapter on FDA their guidance document for radiological imaging and the sources of errors mentioned there
- 2021-11-01: Chapter with FDA's Guiding Principles added
- 2021-09-10: Added link with the statement for the EU
- 2021-07-31: In section 1.b.ii) in the table, added two rows with additional criticisms. Added one paragraph each in the line with definitions
- 2021-07-26: Moved section 2.n) to section 1.b). Added critique and call to action there
- 2021-04-27: Section on EU planning on new AI regulation added

Author:
Daniel Reinsch

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.