In Vitro Diagnostic Medical Device Performance Evaluation: 8 Steps to Conformity
Tuesday, May 5, 2020
If manufacturers don’t conduct a legally compliant performance evaluation of their in vitro diagnostic medical device (IVD), they aren’t just running the risk of problems during the authorization process.
They are risking patient safety. Therefore, the IVDR sets strict requirements for this performance evaluation.
Find out how the requirements of the IVDR for the performance evaluation differ from those of the IVDD to make sure you achieve conformity as quickly as possible.
The most common mistakes can be avoided in eight steps.
1. Why the IVD performance evaluation is so important
Summary
In this section, you will learn about the potential negative consequences of a poor performance evaluation. As an IVD manufacturer, you should make absolutely sure to avoid them.
a) The role of in vitro diagnostic medical devices
The purpose of in vitro diagnostic medical devices (IVDs) is to provide information from human samples, such as blood and tissue, that allows conclusions to be drawn about, for example, physiological or pathological processes in the body.
An IVD is used to identify, for example:
- Tumor markers in blood
- Coronavirus (SARS-CoV-2) in a smear
- Cancer cells in a biopsy
This information is crucial for making a diagnosis and, therefore, for the further treatment of a patient. The IVD performance evaluation must ensure that the information provided is correct and accurate, and that it provides the intended clinical benefit.
b) Potential IVD errors
However, the information generated using an IVD can be incorrect in several respects:
- It is incorrect.
- False positive: A laboratory test incorrectly says that a test result is abnormal or detects a disease even though the patient is not ill.
- False negative: A laboratory test incorrectly says that a test result is not abnormal or does not detect a disease even though the patient is ill.
- The information is inaccurate.
- The IVD does not display the information at all or only displays it with a delay.
There are numerous reasons for such errors. Examples are:
- The analysis procedure is unsuitable in general.
- The manufacturer has not considered all the factors that could affect the tissue samples, e.g., the effect of drugs.
- Reagents change, for example, under the influence of heat, oxygen or light.
- Software errors cause patients to get mixed up.
- When the samples are opened, even the smallest splashes lead to cross-contamination of other samples.
c) Consequences for patients
There is a risk that incorrect information could have far-reaching consequences for patients.
False positive results generally trigger an unnecessary cascade of measures that harm the patient, or at least cause them unnecessary stress:
- Unnecessary blood draws
- Avoidable biopsies
- Wrong therapies, e.g., with medicines or even operations
False negative results, in contrast can lead to critical delays to urgently needed therapies or even have an impact on public health.
d) Consequences for manufacturers
The performance evaluation doesn’t just enable IVD manufacturers to ensure that the risks for patients have been minimized, it also enables them to ensure that the consequences for their own company have been minimized as well.
- The device “fails the authorization” and is therefore released onto the market later. As a result, the company losesplanned sales.
- The device has to be withdrawn from the market. This doesn’t just have financial implications for the company, it also damages their reputation. Manufacturers have to publish such recalls on the websites of the corresponding authorities.
- In the worst case, manufacturers will be faced with claims for compensation from patients harmed by the device.
- In addition to unsatisfied customers and a damaged reputation, manufacturers will also face additional costs for support and improvements.
2. What a performance evaluation is and what it has to demonstrate
Summary
In this section, you will be introduced to important concepts and definitions that you will need to know in order to understand the legal texts.
a) Definitions
Fortunately, the IVDR contains several definitions:
Definition: Performance evaluation
“Assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device”
IVDR Article 2(44)
This definition uses other defined terms:
- Scientific validity
- Analytical performance
- Clinical performance
Definition: Scientific validity of an analyte
“Association of an analyte with a clinical condition or a physiological state”
IVDR Article 2(38)
The basis for each IVD is the analyte that is used to identify a physiological state or disease. For example, there is a link between the coronavirus (SARS-CoV-2) and the disease Covid-19. Similarly, there is a link between the level of prostate-specific antigen (PSA) in the blood and the risk of a man suffering from prostate cancer.
Definition: Analytical performance
“Means the ability of a device to correctly detect or measure a particular analyte”
IVDR Article 2(40)
To ensure that the analyte can also be reliably measured using the method chosen, IVD manufacturers should check the analytical performance of their device. To continue with the examples: The analytical performance of an IVD is its ability to identify the coronavirus as accurately as possible (i.e., without false-positive or false-negative results) or to determine the PSA concentration as accurately as possible.
Definition: Performance of a device
“Ability of a device to achieve its intended purpose as claimed by the manufacturer. It consists of the analytical and, where applicable, the clinical performance supporting that intended purpose”
IVDR Article 2(39)
This definition uses another term, “clinical performance,” that we need to understand.
Definition: Clinical performance
“Ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user”
IVDR Article 2(41)
b) Clinical evidence
IVD manufacturers must provide proof of an IVD’s clinical benefit based on data on scientific validity, analytical performance and clinical performance. Scientific validity refers to the association between the analyte and the disease or physiological state. After all, for the IVD to deliver clinical performance, it must have a reliable and precise method for measuring the analyte. Such a method demonstrates the ability of the IVD to determine clinical, physiological and pathological states or identify processes, and is therefore a vital aspect of the device's performance.
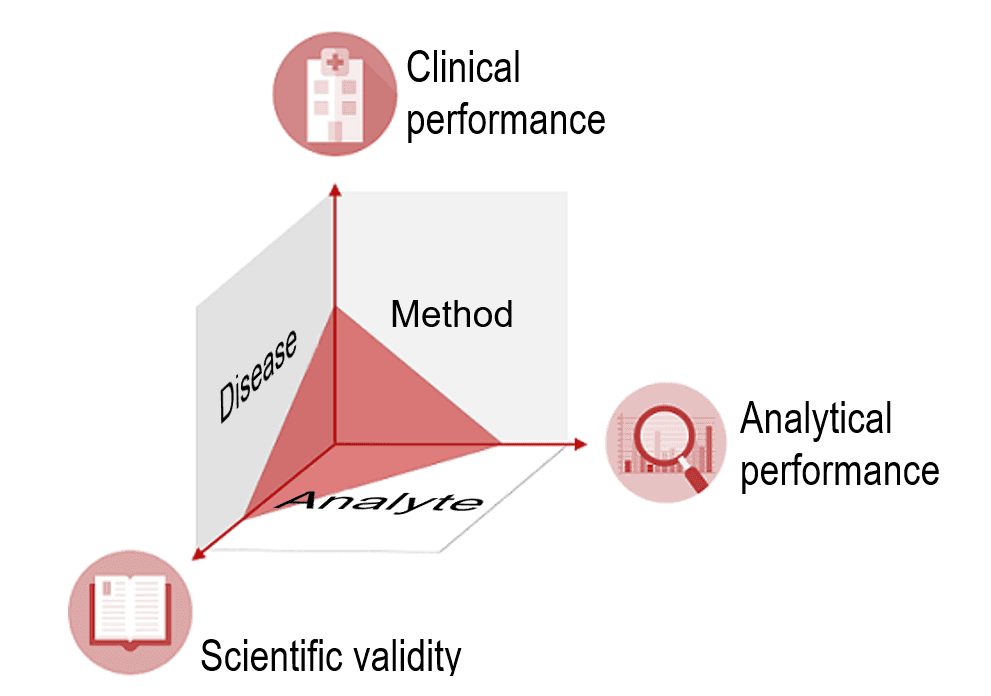
It is also important to note that clinical performance does not refer to an absolute ability but, in fact, depends on the intended purpose and thus the “target population” and “intended user.”
The clinical performance of an IVD may be good for “normal” patients but not for patients undergoing chemotherapy because the accuracy of its measurement is affected by cytostatics.
A device's performance may be excellent for professional users, but not for laypersons. Therefore, the IVDR stresses that an IVD’s performance depends on its intended purpose.
The clinical evidence, i.e., the proof that a device is safe and achieves the intended clinical benefit, is provided by the performance evaluation. This performance evaluation assesses clinical performance, analytical performance, and scientific validity.
3. Which performance evaluation regulatory requirements do IVD manufacturers have to comply with?
Summary
This section will give you an overview of
- The relevant regulations (without having to research them first)
- The most important requirements
You will also find out what deltas there are between the IVDD and IVDR that you may need to close.
Both the IVD Directive 98/79/EC (IVDD) and the IVD Regulation 2017/746 (IVDR) establish, in their respective Annex Is, that the general safety and performance requirements must be complied with. Demonstrating this compliance is part of the performance evaluation.
a) IVDR
Article 5
The IVDR states that in order for a device to be placed on the market or put into service:
“A device shall meet the general safety and performance requirements set out in Annex I which apply to it, taking into account its intended purpose. [...] Demonstration of conformity with the general safety and performance requirements shall include a performance evaluation.”
Article 5, IVDR
Article 56
Article 56 requires manufacturers to confirm that the general safety and performance requirements according to Annex I have been met. Manufacturers also have to carry out a performance evaluation to provide this confirmation.
This performance evaluation includes (as outlined above) the demonstration of the scientific validity of the analyte, the analytical performance and the clinical performance.
The focus is on demonstrating the following in particular:
- The general requirements set out in Annex I Chapter I
- The performance characteristics set out in Annex I, paragraph 9
- An acceptable benefit-risk ratio
The article refers to the requirements established in Annex XIII, Part A with regard to the performance evaluation.
Annexes XIII and ISO 20916:2019
This annex specifies how IVD manufacturers must plan, carry out and document the performance evaluation. Manufacturers should also refer to ISO 20916:2019, which the amended IVDR of May 03, 2019 directly references.
“The rules on performance studies should be in line with well-established international guidance in this field, such as the international standard ISO 20916 on clinical performance studies using specimens from human subjects, currently under development, so as to make it easier for the results of performance studies ...”
IVDR, Recital 66
ISO 20916:2019 was published in May 2019. Its title is “In vitro diagnostic medical devices – Clinical performance studies using specimens from human subjects – Good study practice.”
ISO 20916:2019 describes in detail the requirements for the planning and performance of clinical performance studies. It supplements the IVDR with regard to the description of the organization, the roles involved and the requirements for conducting such a study.
There are overlaps with the IVDR, particular with regard to the content of the clinical performance study plan (according to IVDR, Annex XIII, paragraph 2.3.2). ISO 20916 calls this plan the “clinical performance study protocol (CPSP).”
Annexes A to F of ISO 20916:2019 are dedicated to the specific requirements for interventional and other types of performance studies in accordance with Article 57 of the IVDR. The IVDR itself provides detailed specifications for these studies in Annex XIV.
Annex II: Technical documentation
Annex II describes the technical documentation requirements. In paragraph 6 of this annex, the IVDR describes what the performance evaluation, verification and validation should contain.
Annex VII: A look behind the facade
Annex VII describes the requirements to be met by notified bodies. Understanding what notified bodies have to look for during conformity assessment activities helps us to work out what is important in the performance evaluation and where the interfaces are.
Focus of the assessments
According to Annex VII, paragraph 4.5.4 of the IVDR, the focus of the assessment should be on a manufacturer’s procedures and the documentation regarding:
- The planning, conduct, assessment, reporting and updating of the performance evaluation
- Post-market surveillance and post-market performance follow up (PMPF)
- The interface with the risk management process
- The appraisal and analysis of the available data and its relevance with regard to demonstrating conformity with the general safety and performance requirements set out in Annex I of the IVDR
- The performance evaluation report
The annex explicitly requires notified bodies to take into consideration the available common specifications, guidance and best practice documents.
Literature search
They must also assess the results of the literature searches, of the verifications and validations performed, and of other tests. Notified bodies must also assess the packaging, stability studies, and results of the shelf life tests.
The assessment of the performance evaluation must expressly cover:
- The intended purpose and intended use
- The planning of the performance evaluation
- The methodology for the literature search and its documentation
- The analytical and clinical performance studies
- The PMPF
- The performance evaluation report
- Justifications in relation to non-performance of performance studies or deviations from the specified process
- If data from devices considered equivalent are used for the performance evaluation, the validity of the assumed equivalence and the suitability of the data for demonstrating conformity must be verified
It is noteworthy that the notified body must ensure:
- “That the conclusions drawn by the manufacturer are valid in the light of the approved performance study plan”
- That “the performance evaluation adequately addresses the relevant safety and performance requirements provided for in Annex I”
- That the performance evaluation is “appropriately aligned with the risk management requirements”
- That the performance evaluation “is conducted in accordance with Annex XIII”
- The performance evaluation “is appropriately reflected in the information provided relating to the device”
This gives us hope that there will be significantly more transparency and comparability in future when it comes to information on an IVD’s performance.
b) MPG, IVDD and EN 13612:2012
The requirements of the MPG (German Medical Devices Act) refer to the In Vitro Diagnostics Directive (IVDD). Here too the focus is on the intended purpose. And here too, IVDs must – taking into account their intended purpose – meet the relevant essential requirements according to Annex I. (see MPG § 6, paragraph 2).
“Evidence of the suitability of in vitro diagnostic medical devices for the specified in-tended purpose should be provided through performance evaluation based on appropriate data. The performance evaluation should be based on:
– data from scientific literature which cover the intended use of the medical device and the techniques involved in its use as well as a written report containing a critical evaluation of these data or
– the results of all performance evaluation studies or other appropriate tests.”
MPG, Section 19, paragraph 2
In Annex I, paragraph 3 the IVDD lists the essential performance parameters that an IVD must meet (unless the parameters can be excluded with good reason). Furthermore, in Annex I, paragraphs 1 and 2, the directive describes the requirements for the safety of the device and the acceptability of the benefit-risk ratio.
In the third paragraph of Annex III, indent 11 requires “adequate performance evaluation data showing the performances claimed by the manufacturer and supported by a reference measurement system.”
The standard EN 13612:2002 is harmonized under the IVDD. The standard, which is entitled “Performance evaluation of in vitro diagnostic medical devices” specifies “the responsibilities and general requirements for the planning, conduct, assessment and documentation of a performance evaluation study.”
c) IMDRF/GHTF
Guidance documents reflect the state of the art
Further information of conducting and documenting performance evaluations can be found in the guidance documents from the IMDRF(formerly the GHTF). They represent the current state of the art under the IVDD and were the basis for the procedure as it is now described in the IVDR.
In some places you can find word-for-word the same requirements. Therefore, the requirements of the IVDR are – contrary to what a lot of manufacturers have claimed – not new at all, rather they have been the state of the art since 2012.
Applicable documents
Manufacturers should pay particular attention to the following documents:
- GHTF/SG5/N6:2012 “Clinical Evidence for IVD medical devices – Key Definitions and Concepts”
Since 2012, this document has defined the terms that we now find in the IVDR. This includes, e.g., clinical evidence, scientific validity, analytical performance, clinical performance, and clinical utility (clinical benefit). Section 4.10 even provides an example of how to provide clinical evidence for an IVD. - GHTF/SG5/N7:2012 “Clinical Evidence for IVD medical devices – Scientific Validity Determination and Performance Evaluation”
This document looks at the underlying process for presenting clinical evidence in more detail. The procedures for determining the scientific validity of the analyte, verifying analytical performance and validating the clinical performance are described step by step. The requirements for a systematic literature search (see Annex B) and documenting in it a literature search report (see Appendix A) are also described. - GHTF/SG5/N8:2012 “Clinical Evidence for IVD Medical Devices – Clinical Performance Studies for In Vitro Diagnostic Medical Devices”
This guidance document now goes into detail on the requirements for IVD clinical performance studies. It describes the different types of studies and explains the numerous that have to be considered when planning a clinical performance study. Further chapters outline the content of the CPSP and CPSR and specify the requirements for the conduct of the clinical performance study.
4. A performance evaluation in 8 steps – what you should do as manufacturer
Summary
In this section, we will explain step-by-step how you can conduct target-oriented legally compliant performance evaluations.
Step 1: Create and review a standard operating procedure
First write a standard operating procedure for IVD performance evaluations and integrate it into your QM system. How to create a standard operating procedure is described below.
Make sure that the standard operating procedure covers the entire IVD performance evaluation process. It should cover the complete life cycle of the device and goes far beyond development.
Note: Although ISO 13485 does not explicitly require a process for the performance evaluation to be defined, it does require, in section 4.2.1 e), all other documentation specified by applicable regulatory requirements. And this requirement is found in the IVDR in Article 56 and Annex XIII, among others. The IVDR describes the performance evaluation as a continuous process. It emphasizes the close interface with risk management (Annex VII, Section 4.5.4 and Annex XIII section 1.1).
Step 2: Write a device-specific performance evaluation plan
The next step is to write a specific performance evaluation plan for each IVD. Make sure that you include all the content required by Annex XIII, paragraph 1.1 of the IVDR in the performance evaluation plan.
In the performance evaluation plan, you should describe the current state of the art, with regard to the medically or diagnostically relevant field as well as in terms of the technology.
Research in guidelines, the scientific literature and standards, and document this literature search clearly.
Please note:
- As an IVD manufacturer, you can derive the device requirements (“Design and development inputs” according to ISO 13485, 7.3.3) and the device design specifications (“Design and development outputs” according to ISO 13485, 7.3.4) from the results of the literature search.
- The research into the state of the art will generate results (e.g., about the benefits and risks of comparable devices) that you will also need for risk management.
- Create the performance evaluation plan at the start of the IVD development stage. It is common practice to continuously add to this plan when new findings become known.
Step 3: Demonstrate scientific validity
Now development can start. Demonstrate the scientific validity of the analyte(s) to be detected or measured by the IVD as early as possible in the development cycle.
Carry out a further systematic literature search. Document this search clearly, e.g., the search strategy, the sources used and the criteria for selecting the data. Statements by professional societies and expert opinions can also be included.
If this evidence is not sufficient, carry out feasibility studies. Their results can be used to prove the scientific validity of the analyte, particularly in the case of novel analytes.
Summarize all the results in a scientific validity report. Add the report to the performance evaluation file, which is itself part of the technical documentation.
Note: Use your literature search report in the continuous process to update the performance evaluation and during the post-market performance follow-up (PMPF).
Step 4: Conduct the analytical performance evaluation
Now you can start with the analytical performance evaluation as part of the IVD verification process. Describe your planned procedure and the concrete experimental design in an analytical performance evaluation plan (verification plan).
If you have a complex IVD system (e.g., consisting of an IVD device, IVD assay and IVD software), plan the verification of
- The individual subsystems
- Their integration
- The overall system
As well as the analysis itself, you should also plan the sampling and sample handling precisely because reproducible and accurate analysis results can only be obtained if the conditions during the sampling and preparation of the samples are controlled.
Then conduct the evaluation according to the plan and finally summarize the results of the analytical performance studies in an analytical performance report. Please also evaluate the results in this report.
Please note:
- The demonstration of analytical performance must always be based on analytical performance studies.
- The analytical performance evaluation, as part of the verification, is used to demonstrate the analytical performance parameters according to Annex I, paragraph 9.1 a) of the IVDR.
Step 5: Conduct the clinical performance evaluation
Next move on to the clinical performance evaluation, which will show the diagnostic accuracy of your IVD.
Please note:
- As a general rule, clinical performance study results are required to assess the clinical performance characteristics. In justified exceptions, you can refer only to scientific literature (e.g., in the case of established, standardized tests and where the equivalence of the device has been demonstrated) or to published experiences gained from routine diagnostic tests (see the article on lab developed tests).
- This basic strategy for demonstrating clinical performance is already described in the performance evaluation plan from step 2.
If you use (supplementary) literature data, document your systematic literature search clearly.
Write another plan for the clinical performance studies – the clinical performance study protocol (CPSP). Make sure that you include the aspects required by Annex XIII, paragraph 2 of the IVDR as well as those required by ISO 20916:2019. You should also make sure that the plan provides for the collection of data for all parameters listed in Annex I, paragraph 9.1 b).
Finally, summarize the results of the clinical performance evaluation of your IVD in a clinical performance report.
Step 6: Demonstrate the stability of the IVD
Depending on the type of device you have, you now need to demonstrate its stability. This evidence is particularly important for IVD assays and in vitro diagnostic reagents.
First plan the stability studies. For this, follow the advice in the EP25 guidance document from the Clinical and Laboratory Standards Institute (CLSI). When preparing your plan, make sure that you take all stability aspects mentioned by the IVDR in Annex II, paragraph 6.3 into account. These are in particular:
- The device’s shelf life
- The in-use stability (e.g., of the reagents)
- Transport stability
Step 7: Prepare the clinical performance evaluation report
Lastly, summarize all the results on scientific validity, on analytical performance and clinical performance in the performance evaluation report.
In the report, you should evaluate the clinical evidence in the light of the current state of the art in medicine and demonstrate your device's positive benefit-risk ratio.
Step 8: Create and implement a PMPF plan
Create a plan for the post-market performance follow-up plan (PMPF plan). You should take into account the requirements of Annex XIII, Part B, paragraph 5.2 when doing so.
Note: PMPF is the acronym of the English term “post-market performance follow-up”.
Conduct this follow-up for your CE-marked and marketed device and summarize the results in the PMPF report. Use the results of this report to update the performance evaluation report.
Note: The performance evaluation is a continuous process. The performance evaluation activities are carried out during the device's entire life cycle. Therefore, manufacturers are obliged to continuously surveil their device and update the performance evaluation with new information and data that becomes available.
5. What are the common mistakes made during the performance evaluation?
Summary
This section will describe the biggest mistakes you should avoid. Use this list as a checklist to avoid unnecessary hassle and costs.
a) Unspecific intended purpose
Manufacturers often formulate the intended use in a more general and less specific way:
- No limitation of indication (all types of cancer)
- No limitation of target population
As a result, the performance also has to be demonstrated with no limitations on, e.g., indications and target populations. The scope of the studies required grows exponentially. Furthermore, requirements that apply, for example, for performance studies in children, pregnant or breastfeeding women (see Articles 61 and 62) are easily overlooked.
b) Unsystematically researched or documented state of the art
If manufacturers do not systematically research the state of the art and do not clearly document this research, there will be a lack of
- Reliable specifications of analytical and clinical performance data
- Knowledge and analysis of competitor devices
- User knowledge, the use environment and the intended clinical benefit (what does the physician or patient do when they have the information from the IVD? What actions and therapies are possible?)
Lack of knowledge of the state of the art leads to manufacturers not setting device requirements and, for example, not specifying sufficient controls for the device (e.g., self-tests).
c) No documented systematic literature review
If the literature review is not conducted in full and documented clearly, the manufacturer cannot demonstrate that they have met the performance requirements. Authorities and notified bodies will notice this and refuse authorization.
d) Inadequate plans for analytical performance evaluation
The typical mistakes include missing or incomplete performance evaluation plans. This leads, for example, to analytical performance studies without a statistically valid experimental design.
Reasons for inadequate plans include a lack of knowledge of the state of the art with regard to planning, which is described in the CLSI standards. Incidentally, the IVDD harmonized standard EN ISO 18113-1:2011 “In vitro diagnostic medical devices – Information supplied by the manufacturer (labelling) – Part 1: Terms, definitions and general requirements” refers to these CLSI standards.
e) Unsuitable performance studies
The following mistakes are often observed during clinical performance studies:
- There is no explanation of the number of cases or the number of samples.
- The performance study does not take the intended purpose into account sufficiently. As a result, the study design is not suitable.
- The manufacturer does not derive the clinical performance parameters from the state of the art specifically for the promised clinical benefit. As a result, the performance studies do not have the correct endpoints.
f) No performance data from studies
Manufacturers often hope to get by without clinical performance data from studies. This is possible in principle. However, it requires equivalence between the IVD to be evaluated and the comparator device. This technical and medical or diagnostic equivalence must be analyzed in detail and the evidence documented. Unfortunately, the information required is often missing.
g) Lack of proof of scientific validity
It is not enough to just demonstrate analytical performance. Manufacturers must also prove the scientific validity of the analyte. This is forgotten because the IVDD does not explicitly require this, even though it is the state of the art according to GHTF/SG5/N7:2012.
6. Support from the Johner Institute
The IVD experts at the Johner Institute can provide support to IVD manufacturers with all performance evaluation activities and phases:
- Strategy development
- Determination of the state of the art
- Planning and conduct of the evaluations of
- Scientific validity
- Analytical performance
- Clinical performance
- Planning and conduct of the PMPF activities and studies, and updating of the performance evaluation
The Johner Institute’s IVD team can
- Help you create standard operating procedures for the performance evaluation
- Carry out the literature search for you and help you make sure your documentation is clear and comprehensible
- Create performance evaluation plans and reports for you
- Provide support for all types of study
- Review and, if requested, correct your files before submission
- Hold on-site or online seminars, training courses and workshops
Get in touch if you want to conduct a quick and legally compliant performance evaluation that will be waved through by your notified body with no problems.
7. Conclusion & summary
a) Performance evaluation aspects
The performance evaluation is as important for IVD manufacturers as the clinical evaluation is for medical device manufacturers. Both are based on data that are already available in the literature or that have to be collected through studies.
The nature of the studies and the data differ considerably. When evaluating the performance of IVDs, manufacturers must demonstrate:
- Scientific validity
- Analytical performance
- Clinical performance
Each of these aspects requires its own plans, data and evaluations.
b) Comparison of IVDD and IVDR
The performance evaluation requirements established in the IVDR and the standard ISO 20916:2019 it references are significantly more wide-ranging than those established by the IVDD. However, under the IVDD, the GHTF and CLSI documents and EN 13612:2002 were already considered the state of the art. So, there aren’t any significant differences.
This means that IVD manufacturers who already comply with the relevant regulations are well prepared for the IVDR. However, a lot of IVDs will be subject to a conformity assessment by a notified body for the first time under the IVDR. All IVD manufacturers should prepare well for the detailed review of the performance evaluation file.
c) MDR and IVDR challenge?
The situation is challenging for manufacturers who must comply with the IVDR’s requirements for the performance evaluation and the MDR's requirements for the clinical evaluation because their devices are subject to both regulations. However, this can be avoided with a clever regulatory strategy.
Do you have any questions about this article? Contact us via our micro-consulting team. The Johner Institute’s IVD team will be happy to help.

Author:
Dr. Catharina Bertram

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.