Clinical Data for Medical Devices
Medical device manufacturers are obliged to systematically collect and evaluate clinical data, both before and after the approval of their products.
The EU Medical Devices Regulation (MDR) increases the requirements for the scope and quality of the required clinical data.
This article provides you with an overview of the regulatory requirements and gives tips how to implement them.
Many manufacturers are not aware of the importance of clinical data in demonstrating the compliance of their products. This concerns both the approval of the products and the "post-market activities", especially the "post-market clinical follow-up".
Tip
Collect the necessary clinical data now, while your products are still approved under MDD. Contact us to find out how you should proceed to avoid costly clinical trials.
1. Clinical data: Basics
a) Definition
Similar to MEDDEV 2.7/1, the Medical Devices Regulation MDR defines the term “clinical data” as follows:
Definition: “Clinical data”
“’clinical data’ means information concerning safety or performance that is generated from the use of a device and is sourced from the following:
- clinical investigation(s) of the device concerned,
- clinical investigation(s) or other studies reported in scientific literature, of a device for which equivalence to the device in question can be demonstrated,
- reports published in peer reviewed scientific literature on other clinical experience of either the device in question or a device for which equivalence to the device in question can be demonstrated,
- clinically relevant information coming from post-market surveillance, in particular the post-market clinical follow-up;”
b) Purposes to be achieved with clinical data
This definition includes both purpose and source of clinical data.
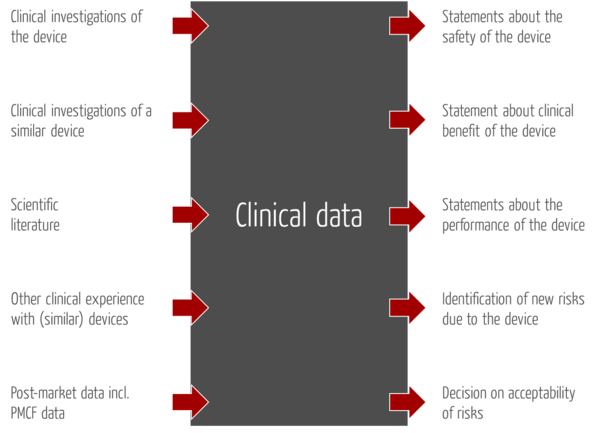
Annex VI reveals that the term “performance” is interpreted in two ways:
- Performance in the sense of a capability in the form of specified performance parameters. An example is the ability of a defibrillator to deliver a certain amount of energy per unit of time.
- Performance in the sense of clinical benefit and achievement of the intended purpose of the product. In the case of the defibrillator, this would be to terminate ventricular fibrillation.
The proof that the specified performance claims (1st point) are fulfilled is usually provided by manufacturers based on preclinical data. Examples of such data can be found below.
In Annex XVI, the MDR discloses further purposes of clinical data:
- They are intended to identify previously unrecognised risks/hazards and to reassess risks.
- Based on updated clinical data, manufacturers are to reassess their decision on the acceptability of risks.
2. Sources of data
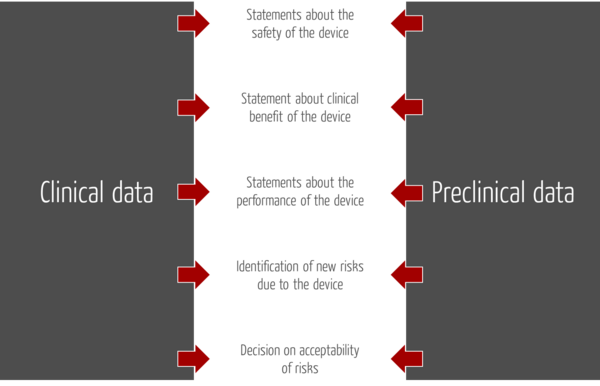
In the context of the clinical evaluation, manufacturers should not only consider clinical data, but also preclinical data.
a) Sources of clinical data
Data | Examples |
Clinical trials with the product | Clinical trials Application observations Internal (unpublished) clinical data e.g. case series (level 5 studies) |
Clinical trials with the equivalent product | Clinical trials published in the scientific literature Internal (unpublished) clinical data e.g. case series (level 5 studies) with a predecessor product |
Scientific literature | Cochrane standards |
Other clinical experience with (equivalent) products | User reports Official databases (BfArM, MAUDE) |
Post-market data including PMCF data (clinical follow-up) | Error reports, customer complaints Calls to hotline Service technician reports User observations Clinical studies (clinical follow-up) User reports Regulatory databases (BfArM, MAUDE) |
b) Sources for preclinical data
Examples of preclinical data include the results of the following tests
- Biocompatibility
- Electrical and mechanical safety e.g. according to IEC 60601-1
- Electromagnetic compatibility e.g. according to IEC 60601-1-2
- Software tests (unit, integration and system tests)
- Summative assessment of suitability for use
- Durability and stability (e.g. during long storage, transport, heat, cold)
- Animal tests
- Simulations
- Laboratory tests
Data in this list, such as post-marketing usability tests, can contain clinically relevant information and can be considered to be clinical data.
3.Regulatory Requirements for Clinical Data
a) Europe
The Medical Device Directive MDD, like the Medical Device Regulation MDR, requires that manufacturers collect and evaluate clinical data. In the MDR, these requirements are found in the following chapters:
- Article 61 requires a clinical evaluation based on “sufficient clinical data”. Also: “The clinical evaluation and its documentation shall be updated throughout the life cycle of the device concerned with clinical data […].” The frequency depends on the class of the product and is specified in Article 84 of the MDR.
- Annex II (Technical Documentation) requires pre-clinical and clinical data in Chapter 6 (Verification and Validation of the Product).
- Annex XIV Part A (clinical evaluation) goes into particular detail on clinical data.
- Annex XIV Part B (clinical follow-up, PMCF) requires manufacturers to collect and evaluate clinical data "in a proactive manner".
Conclusion
The MDR requires manufacturers to collect clinical data before and after authorisation. It already specifies in the definition which sources the manufacturers must at least take into account.
According to Annex XIV, manufacturers may only rely on clinical data from other products in the clinical evaluation if the equivalence to these other products is demonstrated. The MDR requires technical, biological and clinical equivalence. Read more about this below.
The importance of clinical data to the MDR is revealed in Annex VII, which specifies the requirements for notified bodies: “The notified body […] shall pay particular attention to clinical data from post-market surveillance and PMCF activities […]”.
b) USA/FDA
The FDA requires clinical data from the manufacturers for the approvals, e.g. the premarket notifications PMN or premarket approvals PMA. In the case of PMNs (510(k)), these usually come from the comparator product, as the FDA itself says.
At the end of February 2018, the FDA published a guidance document that describes in which form the FDA accepts which clinical data. It is entitled "Acceptance of Clinical Data to Support Medical Device Applications and Submissions Frequently Asked Questions".
2023 FDA has published the guidance document "Recommendations for the Use of Clinical Data in Premarket Notification [510(k)] Submissions."
4. Typical Problems
a) Equivalence of products
Most medical device manufacturers aim to avoid clinical trials with their own product. Clinical trials are expensive and usually take months. Therefore, manufacturers try to use the clinical data of comparator products and/or provide evidence according to the "literature procedure".
However, these comparator products must be "sufficiently similar". The MDR and MEDDEV 2.7/1 Revision 4 have significantly increased the requirements for this equivalence. Equivalence has three aspects:
- “Technical: the device is of similar design; is used under similar conditions of use; has similar specifications and properties including physicochemical properties such as intensity of energy, tensile strength, viscosity, surface characteristics, wavelength and software algorithms; uses similar deployment methods, where relevant; has similar principles of operation and critical performance requirements;
- Biological: the device uses the same materials or substances in contact with the same human tissues or body fluids for a similar kind and duration of contact and similar release characteristics of substances, including degradation products and leachables;
- Clinical: the device is used for the same clinical condition or purpose, including similar severity and stage of disease, at the same site in the body, in a similar population, including as regards age, anatomy and physiology; has the same kind of user; has similar relevant critical performance in view of the expected clinical effect for a specific intended purpose.”
[Source: MDR]
The Johner Institute regularly experiences that notified bodies interpret the equivalence requirements so highly that clinical trials become unavoidable. This is not always comprehensible or sensible, especially in the case of non-critical products.
b) Quantity and Quality of Data
According to MDR, data must be “[…] of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s) […]”. For this, the data must be “scientifically valid, reliable and robust”.
The Notified Body assessors place particular emphasis on the following characteristics:
- The clinical data must be quantitatively sufficient to demonstrate statistical significance.
- They must come from sources whose scientific validity does not raise doubts.
- The data from different sources should support the manufacturer's thesis and thus provide evidence of the product's benefit, performance and safety.
- As far as possible, all sources must be searched or evaluated. Some manufacturers resist the requirement to use the EMBASE database, for which a fee is charged. There is no requirement to use all data sources. Pubmed is recognised as the sole source by most notified bodies.
- As mentioned above, the data must have been obtained with products, technologies or procedures for which the requirements for equivalence and thus comparability are met.
Especially the assessment of the scientific validity of clinical data leads to discussions between manufacturers on the one hand and notified bodies and authorities on the other. This involves questions such as:
- Is the scientific journal reputable? The impact factor of the journal is a relevant metric.
- Is the study design appropriate? Do the data have to come from a prospective, randomised and controlled study?
- Does the study have a bias? Did the selection of the subjects or the procedure or even the manufacturer itself have an influence on the results?
The GRADE, a "level of evidence" according to Cochrane, attempts to quantify this scientific quality of publications.
c) Obligation for Clinical Trials
Especially for class IIb implants and all class III devices, the MDR insists that manufacturers (also) collect clinical data in the context of clinical trials with the specific device. Exemptions exist, but the number of clinical trials will increase.
5. Tips and Conclusion
The demands on manufacturers to scientifically prove the benefit and safety of their products based on clinical data are increasing. However, the collection of scientific data, e.g. in the form of a clinical trial, must not become an end in itself. We therefore recommend:
a) Use performance data (if possible)
The EU directives or regulations allow (MDD Annex X, Paragraph 1.1d or Chapter VI Article 61 Paragraph 10 of the MDR) to use preclinical data such as performance data, simulation results and system tests instead of clinical data in certain cases. This should be particularly successful for non-critical products with no (or little) patient interaction. Examples of such products where the Johner Institute team succeeded in following this path are: Stand-alone software, oral spatulas, dental chairs, very established material medical devices and very established cardiac electrodes.
b) Pay attention to the database of clinical trials
Manufacturers must always consider the scientific literature. We also recommend considering registries and clinical trial databases such as https://clinicaltrials.gov/.
c) Use data from predecessor products
Many companies underestimate the value of data derived from predecessor products. Sometimes it is easier to conduct a non-interventional use study with such a predecessor product than to set up a new clinical trial. Coordinate this approach with your Notified Body.
d) Start collecting the data immediately
With the MDR, you have to re-certify your product. Use the time until then to collect enough data with the already authorised products that you can fall back on later and thus avoid a clinical trial.
The intended purpose is a prerequisite for any clinical evaluation. This purpose statement should specify the intended clinical benefit as quantitatively as possible.
Do you need assistance?
Feel free to contact the Johner Institute if your notified body requires a time-consuming and cost-intensive clinical trial even though you believe you have sufficient clinical data. Our clinical experts regularly find a way out. They will also help you to conduct a clinical trial or observational study quickly, in a plannable, cost-efficient and legally compliant manner.
Change History
- 2023-10-23: Article updated e.g., by reference to new FDA Guidance Document. Chapters 1 and 5 restructured.
- 2018-09-12: First version published

Author:
Dr. Bettina Martin

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.