International Medical Device Authorizations: 5 Steps to New Markets
A lot of medical device manufacturers see the international authorization of their device as a potential hazard: the opportunities offered by new markets are offset by hard-to-calculate risks as well as the time and costs required to obtain these authorizations.
Manufacturers will be better in a better position to manage these risks if they follow these five steps and use the “international authorization” checklist. This is essential because a failed authorization is not the “worst case”.
1. The challenges you have to overcome during international authorizations
In a survey conducted by the Johner Institute, participants told us the most frequent difficulties they encountered when obtaining international authorization of their devices.
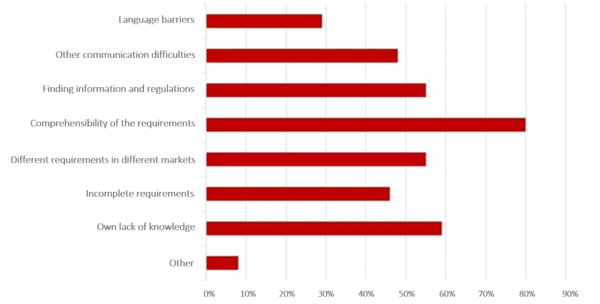
The authorization process often drags on as a result of simple communication difficulties. The reasons for this are complex and range from language barriers, to different understandings of requirements, through to cultural customs.
Finding concrete and correct requirements is also always a hurdle. Regardless of whether it is provided by local distribution partners involved in the authorization or by authorities, the information is often incomplete or only provided bit by bit.
In addition, terms can be defined in different ways, which can lead to misunderstandings. And everyone who has ever gone through an authorization process knows the challenges of bureaucratic structures.
Some even get the feeling that the authorities act arbitrarily if the documentation the manufacturer has gone to great effort to create to the best of their knowledge still does not meet the requirements in the end.
In the survey, a lot of manufacturers also classified their own lack of knowledge as an obstacle. They also frequently mentioned the fact that there are different requirements in different countries as a difficulty.
Conclusion: Going through the international authorization process can be expensive, time-consuming and frustrating.
NB!
Even an authorization that a lot of time and energy was invested in and that still failed is not the worst case. In the 2020-03 podcast, Luca Salvatore, a Johner Institute expert on international authorizations, talks about scenarios that can threaten the existence of the company.
2. 5 steps to international medical device authorizations
To prevent your authorization process from failing to overcome these challenges, the Johner Institute recommends going through the authorization process in five steps. These steps will help you to:
- Plan the international authorization in a structured way
- Avoid unforeseen costs and work
- Minimize risks
A checklist at the end of the article will ensure that you don’t forget anything when drawing up your plans and that no subsequent work that could delay or even jeopardize the authorization is required.
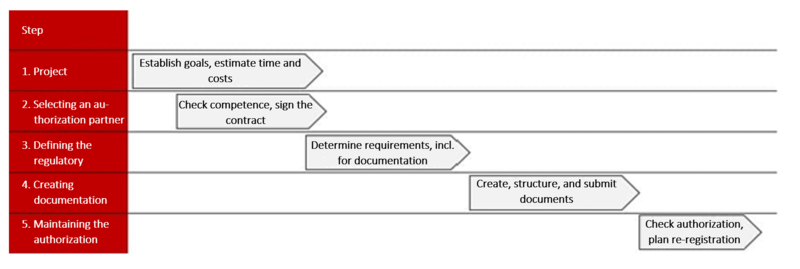
Step 1: Project planning
Every project starts with an objective. This should include:
- Selecting the markets
- Selecting the devices
- Time and sequence of market entry
Identify the authorities that regulate the authorization of your medical device in the country in question. The website of the corresponding authority won’t just contain (most of) the authorization requirements, it will also contain initial information on the process and the costs.
If possible, determine the classification of your device and the corresponding authorization pathway. These aspects usually have an impact on duration and costs.
These costs include the internal costs and regulatory fees and, if applicable, the ongoing costs of maintaining the authorizations. Maintaining an authorization often costs several thousand euros per year.
If you are missing information (e.g., because it is not available in English) that you need to complete the initial project planning, use:
- A local authorized representative who can communication with authorities locally
- An authorization partner who will be responsible for the registration
- A consultancy company that specializes in international authorizations
- The competent authority Particularly in the case of devices that use novel technologies, consulting with the authorities early to agree on the authorization process can be advantageous.
The checklist (linked to below) will help you ensure that you have all the relevant information for planning the project and, therefore, the duration, costs, and time and effort.
Step 2: Selecting an authorization partner
A local authorization partner is mandatory in a lot of countries. This person must, for example, carry out the registration and authorization, and communicate with the authorities as an authorized representative.
Tip
Choose your authorization partner carefully, since you often become dependent on them as the authorization is in their name.
Options
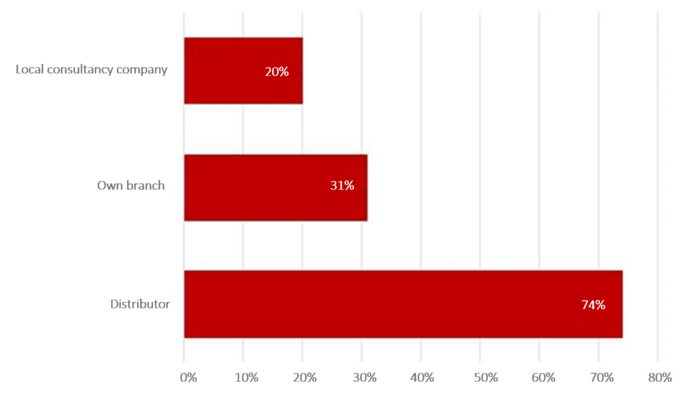
As a rule, a distribution partner, an independent company or a branch in the target country make suitable authorization partners.
In the Johner Institute's survey, over 70% said that the distribution partner was their main contact for authorizations. Branches of the same company follow in second place (31%). 20% of companies work with an independent company.
Advantages and disadvantages
Each type of authorization partner has advantages and disadvantages:
| Advantages | Disadvantages |
Distribution partner | + Possible room for maneuver when negotiating the authorization costs + Single contact in target country + Distribution partner already knows the device | – The authorization is (often) in the name of the distribution partner This means you are dependent on them. – The representative (in this case, your distribution partner) can name distributors in some countries Your distribution partner might not do this. – A change of distribution partner often requires a new registration or a change of registration – Knowledge of regulatory requirements may be limited |
Independent company | + Being independent of the distribution partner + Cooperate with experts in medical device authorizations + Can name distribution partners without changing the authorization | – Potentially higher costs – You also need a distribution partner |
Own branch | + Independent management of authorizations + No additional fees for license holders + Can communicate directly with the authorities | – Barely any disadvantages in terms of the authorization. Sometimes it can be difficult to find qualified staff with experience in medical device authorizations in the target country |
Table 1: Advantages and disadvantages of different types of authorization partners
Decision criteria
Before deciding which authorization partner is best for you, ask yourself the following questions:
- How much time and effort does changing authorization partner require? Is it even possible?
- Is having several distributors for one device permitted?
- Who is allowed to appoint more distribution partners?
- Is it possible to have a device authorized through several partners?
If you already have a partner you want to use, check whether they meet the necessary requirements for the authorization of the device. Important requirements are:
- Existence of a quality management system
- Expertise
- Regulatory approvals
- Reliability (if necessary, request references)
Content of the contract
The contract with your authorization partner should:
- Ensure you get full access to the authorization documentation.
- Ensure you get access to the certificate or a copy of the certificate.
- Cover market surveillance obligations and how customer feedback is handled
- Cover initial and ongoing costs for the authorization, communication with the authorities, feedback, etc.
Tip
Your authorization partner is a supplier you have to evaluate according to ISO 13485:2016.
Step 3: Defining the regulatory framework
You now need to establish the detailed requirements for you as a manufacturer and for the device itself. The specifications are often available as laws, as guidance documents, as referenced standards as well as information on official websites.
If necessary, get help from your authorization partner, the authorities or a consultancy company.
Tip
Country-specific information can also be found in our other articles and in our overview of regulatory authorities.
Documentation requirements
Above all, find out:
- Which documents you have to submit to the authorities
- The required content for these documents
- What form and what language the submission should be made in
Please note that not all countries accept English. For user and service manuals, in particular, countries generally insist on translations into the local language.
Tip
Use qualified and certified translation service providers. Translate critical elements back into German or English if necessary. It is often the case that submission documents have not been correctly translated.
Technical requirements for international authorization
It is not uncommon for compliance with country-specific versions of standards to be required. If you know these requirements at an early stage, you can test these requirements, e.g. for IEC 60601-1, at the same time as your regular testing.
Some countries also require additional testing for certain device groups. More detailed research with regard to the following points is often worthwhile:
- Requirements for radio modules
- Requirements for electronic devices
- Novel technologies (e.g., recognition of clinical trials“)
Step 4: Creating documentation
Complete technical documentation is required for a quick and smooth authorization. Although almost every country has its own requirements, there are some common elements, such as:
- Proof of your quality management system
- Device description, including variants
- A clearly defined intended purpose
- Results of the risk analysis
- Instructions for use and labeling
- Results of the device testing, including software verification and validation.
Use the internationally accepted document structures, such as STED, which the ASEAN member states are looking for with the Common Submission Technical Documentation (CSTD).
An alternative is the ToC (table of contents) format from the IMDRF, which is what Canada and China are looking for. The ToC format also covers large parts of the technical documentation required by the MDR.
This gives you a base you can use to extract the necessary information more easily.
Your or your authorization partner submit this documentation to the corresponding authority. If the submission is successful. you will receive confirmation, e.g., in the form of a certificate.
You should first check that the certificate has been issued correctly and that the manufacturer and device information is correct. If an expiry date is given, clarify the format used for this date. It is not uncommon for the month to be given first before the day.
Step 5: Maintaining the authorization
The motto “after the authorization is before the authorization” applies. Therefore, you should determine how much time you need to plan for re-registration.
In addition, a lot of countries expect the authorization to be updated if any modifications are made to the device. Keep a record of when you have to send a modification notification to the authorities.
In most countries, post-market surveillance is an additional requirement for maintaining an authorization. ISO 13485:2016 also requires you to surveil the requirements in countries where you are selling the device.
Determine the requirements for the reporting of incidents and recalls and establish – together with your local partners – corresponding systems.
3. “International authorization” checklist
Each of the above steps requires a set of activities and results. The international authorization checklist helps you:
- Ensure you do not overlook any necessary activities
- Avoid corrections and thus project delays
- Increase regulatory security
- Make the planning of international authorizations easier
International authorization checklist V01Download
4. Conclusion
The international authorization of medical devices poses countless challenges. But good planning helps to identify and overcome these challenges.
A lot of markets are worth the trouble of international authorization. The five steps and the checklist help control the risks as well as the time and effort required for these authorizations.
What are your experiences or the biggest challenges you had to master? We want to hear your experiences of entering the market in new countries, e.g., either in the comment field or by email.
Do you have any questions on international authorization procedures? The Johner Institute's team will be happy to support you every step of the way and, if required, will also take care of the complete authorization process for you. Contact us now.

Author:
Margret Seidenfaden
Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.