Post-Market Surveillance Plan: Complying with the Requirements of ISO TR 20416
With the post-market surveillance plan, manufacturers are trying to achieve two (potentially) contradictory goals. On the one hand, they have to comply with the legal requirements and maximize the safety of their devices. And on the other, they can only take on as much work as they can realistically do in the long term.
But can this balancing act succeed? Does ISO 20416 help at all?
Find out what the 7 most common mistakes that medical device manufacturers should avoid are. Particularly because even now they regularly lead to problems in audits.
1. What you should know about the PMS plan
Before you read any further, you should make sure you are familiar with the objectives of post-market surveillance and the post-market surveillance plan.
a) Objectives of post-market surveillance (PMS)
Post-market surveillance is a systematic device-specific process carried out with the aims of:
- Detecting undesirable side-effects and risks from medical devices promptly
- Working out the effects on the device's use environment
- Identifying user problems
- Collecting information on improper use
- Collecting information on other defects
Conclusion: During the post-market surveillance process, manufacturers should look for information that will help them maintain or even improve the safety and performance of their medical device.
Further Information
Read more on the basic principles of the PMS process. This article will also help you better understand the difference between vigilance and the post-market clinical follow-up.
b) Objectives of the post-market surveillance plan
The post-market surveillance plan (PMS plan) is used – as the name suggests – to plan post-market surveillance activities. It must therefore define, on a device-specific level:
- which people and roles
- are responsible at which times and on which occasions
- for which activities,
- which information these people collect and
- how they evaluate this information,
- who they have to communicate this evaluation to and
- which actions or notifications they have to trigger.
Conclusion: The aim of the post-market surveillance plan is to make sure that everyone involved knows what they need to do, when, and how in order to achieve the post-market surveillance objectives: the safety of the devices and therefore of the patients, and ensuring the company’s compliance with all the regulations.
2. Requirements of the MDR regarding post-market surveillance
The MDR establishes the requirements for the post-market surveillance plan in Article 84 and Annex III Section 1.
a) MDR Article 84
Article 84 of the MDR states:
The post-market surveillance system referred to in Article 83 shall be based on a post-market surveillance plan, the requirements for which are set out in Section 1.1 of Annex III...
b) MDR Annex III Section 1
In paragraph (a), Annex III Section 1 lists the requirements for the information sources to be included:
- information concerning serious incidents, including information from PSURs, and field safety corrective actions;
- records referring to non-serious incidents and data on any undesirable side-effects;
- information from trend reporting;
- relevant specialist or technical literature, databases and/or registers;
- information, including feedbacks and complaints, provided by users, distributors and importers; and
- publicly available information about similar medical devices.
In paragraph (b), the MDR establishes the requirements for the methods that manufacturers have to use to evaluate these sources of information, e.g.:
- effective and appropriate methods and processes to assess the collected data;
- effective and appropriate methods and tools to investigate complaints and analyze market-related experience collected in the field…
The requirements of the IVDR with regard to the post-market surveillance plan are essentially identical to those in the MDR.
3. ISO TR 20416: a useful tool?
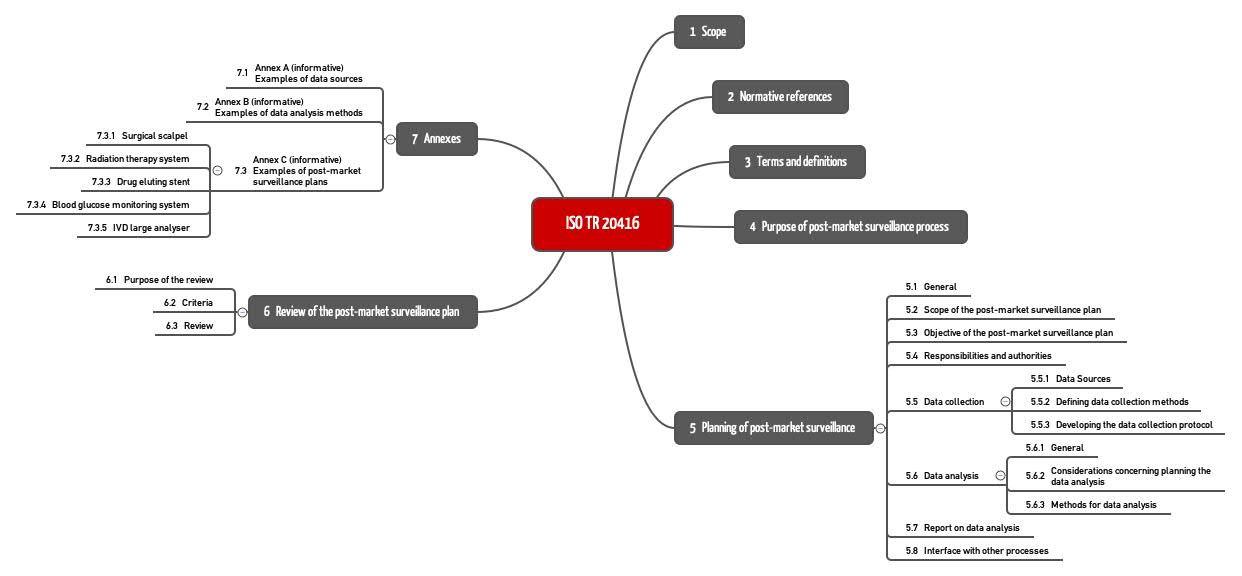
The “Technical Report” ISO TR 20416 is entitled “Medical devices — Post-market surveillance for manufacturers”. This international standard also looks at the post-market surveillance plan.
a) Overview of ISO 20416
In section 4, ISO 20416 states the aims of post-market surveillance and in section 5 it details the requirements for the post-market surveillance plan. Section 6 describes the review of this plan. This examples in Annex C are worth paying attention to.
b) Inter-relationship of ISO 20416 with ISO 13485 and ISO 14971
ISO 20416 also describes the inter-relationship with ISO 13485 and ISO 14971 (see Fig. 2).
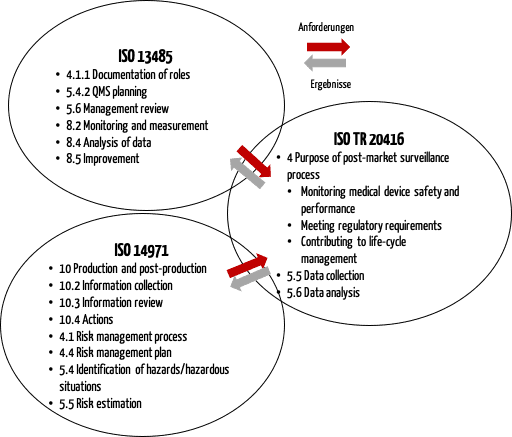
This figure shows that the PMS process must be linked to other quality management processes, particularly with risk management.
ISO 20416 contains specific requirements for manufacturers:
- Ensuring the competence of the responsible roles in the PMS process
- Keeping the regulatory requirements up-to-date (“Regulatory Update”)
- Including the required “input” in the management review and, e.g., defining key figures and reasonable time periods
- Establishing methods for analysis, e.g., procedure for the categorizing or prioritizing of customer feedback
- Continually evaluating and monitoring risks and risk-minimizing measures
4. Post-market surveillance plan section structure (ISO 20416)
a) Overview
ISO TR 20416 proposes a specific section structure for the PMS plan (see Figure 3).
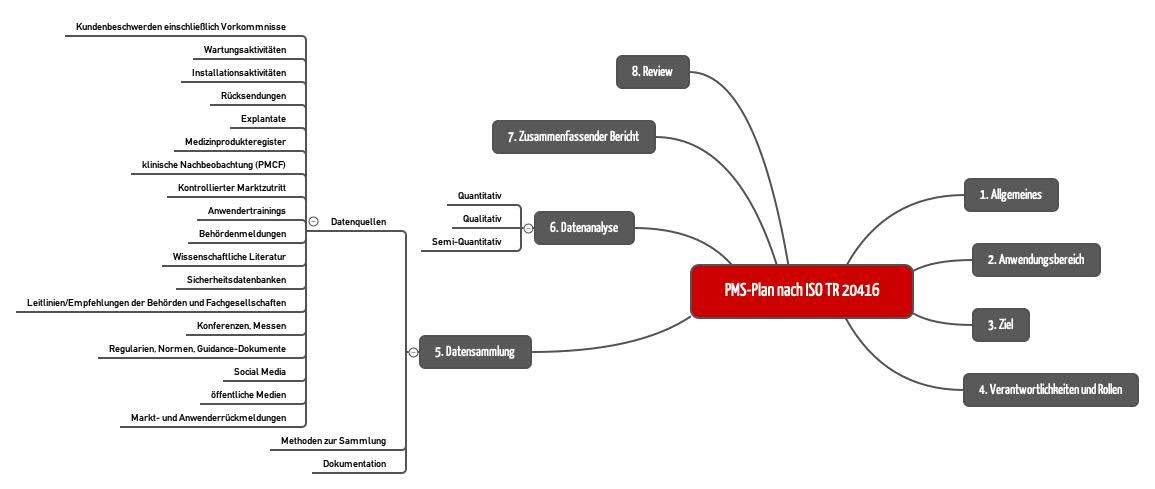
b) Information in the standard on the PMS plan sections
1. General information
The standard does not provide any specific information for this section.
2. Scope of the PMS plan
This should include the:
- Medical device, medical device product family and accessories, and user group
- Classification and markets
- Expected lifetime, expected use figures
- Available clinical data
- Statement on the degree of innovation in the device
3. Objective of the PMS plan
To work out the truly device-specific objectives in addition to the general aims (detection of new risks, compliance with the regulatory requirements), ISO TR 20416 proposes a list of questions and gives some concrete examples:
- Monitoring of the safety and performance at frequent intervals for a limited period of time (new device)
- Ensuring that the links between the clinical evaluation, pre-clinical studies and risk management processes are robust and transparent (new device)
- Obtaining information on the long-term safety and performance of the medical device, including the clinical benefit (device with CE marking after clinical study)
- Confirmation of the prevalence of known or suspected adverse events (device with CE marking after clinical study)
- Monitoring long-term user satisfaction with the medical device and the development of the state of the art (established device)
- Collecting feedback for improvements not necessarily related to safety and performance issues (established device)
4. Responsibilities and authorities
A competent person must be assigned to each activity in the PMS process.
5. Data collection
The standard requires device-specific information sources to be listed and data collection methods to be established. The collection of the data must be documented accordingly.
6. Data analysis
Here the standard distinguishes between
- Quantitative methods (e.g. descriptive statistics, inferential statistics)
- Qualitative methods (e.g., context analysis, narrative analysis)
- Semi-quantitative methods
7. Summary report on data analysis
The plan should give an idea of the report to be created. This includes specifications for the time period and a template to be used.
8. Review of the PMS plan
The plan must be reviewed regularly and, if necessary, modified. Responsibilities, times and criteria should also be defined for this.
c) Conclusion
The PMS plan section structure proposed by ISO TR 20416 is comprehensible and useful.
The standard makes it clear that it is no longer sufficient to say that the support data and the reports to the authorities will be checked once a year for information. It describes numerous other sources of information and their benefits.
What the standard unfortunately does not explain is how individual findings can be evaluated in a reasonable and objective way. And that leads us to the next section.
5. Avoiding the 7 most common mistakes
The Johner Institute doesn’t just carry out post-market surveillance for dozens of medical device manufacturers, it also prepares manufacturers for audits by notified bodies by auditing manufacturers and reviewing their post-market surveillance plans and results. We see the following mistakes again and again:
Mistake 1: not documenting the search strategy
If the PMS plan does not precisely define the search strategy, the manufacturer cannot be sure that two assessors will obtain the same search results.
The plan should, therefore, also specify exactly which search terms, search criteria and search filters the assessors used in which databases and other sources of information.
A template helps to make sure that the PMS plans for all devices contain these precise specifications.
Mistake 2: subjective, non-transparent and undocumented evaluation of the results
Even when the two assessors have the same search results, it is not certain that both will come to the same result in their evaluation. But such consistency is also required.
This means a concrete data evaluation strategy or a precise list of questions and criteria is required. This is the only way to obtain an objective analysis.
In addition, manufacturers must ensure that the analysis is transparent and traceable. To do this, they need to:
- Document the data evaluated, as well as the results and interim results of the evaluation
- Use a template that ensures a uniform structure and comprehensibility
- Refer to the criteria they used to arrive at the result of the analysis
The PMS plan should also include these analysis criteria.
NB!
Establishing these analysis criteria in a higher-level SOP (usually only) makes sense if the manufacturer is surveilling very comparable devices and analyzing the information from such surveillance.
Mistake 3: non-device-specific PMS plans
To obtain really relevant results, the plan must be device specific. For example, it doesn’t make sense to search the FDA databases if the device is not actually regulated as a medical device in the USA.
In contrast, anyone who uses OTS software, such as operating systems and databases, will not be able to avoid the NIST databases even if they don’t sell the device in the USA.
The PMS plans must be specific to, for example, the:
- Device type, device category
- Medical indication and intended purpose
- Intended users, intended use environment
- Technologies and procedures, e.g., production procedures, used
- Markets
- Lifetime, sales volume, frequency of use
Mistake 4: PMS plans not coordinated with other documents
Manufacturers shouldn’t see post-market surveillance plans as isolated documents. Instead these PMS plans must be coordinated with, for example, the:
- Intended purpose
- Clinical evaluation
- Risk management files
The clinical evaluation recommends PMS actions and PMCF actions. Are these in the PMS plan?
The risk management plan defines the monitoring in the downstream phase. Does the PMS plan include these activities?
Mistake 5: no (meaningful) definition of statistical significance
Granted, the requirements of the MDR are difficult to understand and comply with:
“...methods and protocols to be used to establish any statistically significant increase in the frequency or severity of incidents...”
Annex III, Section 1(b), MDR
Statistical significance is often a problem, especially with low numbers of cases. In these situations, you can often only make a case-by-case assessment. ISO TR 20416 explicitly supports such an approach.
Mistake 6: not (sufficiently) defining responsibilities, lack of resources
The PMS is linked to a lot of processes and requires input from several actors. Therefore, a lot of roles have to be involved in PMS and described accordingly in the PMS plan. These include:
- Risk manager
- Safety officer and/or PRRC
- Support
- Service
- Production
- Clinical affairs
- Users
- Distributors
- Suppliers
- Management
These roles and people should know that they are included in the post-market surveillance plan. And, therefore, they should have sufficient capacity and competence.
Mistake 7: not checking information sources at appropriate intervals
A lot of manufacturers assume that an annual review of the information sources is sufficient. But for a lot of devices, this might not be the case.
- New devices
Often with new devices, a lot of questions regarding safety, performance and clinical benefit have not yet been adequately answered. - Devices with rapidly changing technologies and regulations
For example, new documents on new machine learning techniques and "interpretability" are being published every month. The frequency with which new regulations are published also makes an annual review seem inadequate. - Devices with OTS software
IT security databases sometimes publish hundreds of vulnerability reports per day(!).
For these devices, in particular, manufacturers have to surveil the market more closely.
Tip
The PMS experts at the Johner Institute can help you:
- Answer questions on PMS plans
- Quickly review your existing PMS plans
- Write new PMS plans
As a result, they can help you achieve regulatory certainty, and not just avoid problems during audits but also avoid the expense of unnecessary PMS activities. This will also help you ensure the safety of your devices.
Just send us a message here. The Johner Institute’s experts look forward to meeting you!
6. Conclusion, recommendation
Post-market surveillance can and must make a significant contribution to ensuring, and even improving, the safety and performance of medical devices.
a) Importance of the PMS plan, support by ISO 20416
The requirements for effective PMS are a device-specific PMS plan and the provision of the necessary resources to search and evaluate all the relevant information sources.
ISO 20416 helps with a proposal for the structure of the plan. On the other hand, the standard does not help manufacturers with the challenges of evaluating reports. In Annex B, it gives some tips for how manufacturers can identify the trends required by the MDR and set thresholds.
b) The worst is yet to come. Fortunately, there are solutions.
The quantity of data that manufacturers have to collect, process and evaluate for post-market surveillance is continuously increasing. The frequency of this monitoring is becoming shorter, from annually to continuously. This is the case for software-based products in particular. The post-market surveillance plans must define this frequency accordingly.
Manufacturers will only be able to cope with this quantitatively and qualitatively increasing load through automation.
That’s why the Johner Institute has already started to automate these processes. It offers this service to manufacturers to save them considerable time and costs for repetitive work.

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.