Clinical evaluation of software: Three proofs of conformity
The same legal requirements apply to the clinical evaluation of software as to the clinical evaluation of all medical devices. This means that as a Medical Device Software (MDSW) manufacturer, you must prepare a clinical evaluation for your product just like any other manufacturer. A performance evaluation must be carried out for software that is an in vitro diagnostic (IVD) device.
That, in turn, requires clinical data on the device itself, perhaps from a clinical investigation or clinical performance study, or on a proven equivalent product. For software, this approach often makes little sense.
However, the MDCG document MDCG 2020-1: Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software shows a possible alternative path for software products. The Notified Bodies have now accepted the first clinical evaluations of software prepared by us on the basis of this document.
1. Quick recap: What is a clinical evaluation or a performance evaluation?
Definition: Clinical evaluation of a medical device
“Clinical evaluation means a systematic and planned process to continuously generate, collect, analyze and assess the clinical data pertaining to a device in order to verify the safety and performance, including clinical benefits, of the device when used as intended by the manufacturer”
Source: MDR Article 2
Definition: Performance evaluation of an IVD
“Performance evaluation means an assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device”
Source: IVDR article 2
The goals are mentioned, albeit indirectly, right here: It is about proving whether the medical device
- provides the benefit promised by the manufacturer,
- is safe and does not pose any risks that have not already been identified and assessed as acceptable in the risk analysis, and
performs as promised by the manufacturer.
Additional information
Read more articles here about clinical evaluation and about the performance evaluation of IVDs.
2. Clinical evaluation of software: Special features
a) The essential difference: The interfaces
While other medical devices have many forms of “outputs” (= interfaces to the outside world), such as electrical energy (e.g., RF surgical device), electromagnetic energy (e.g., CT) or mechanical energy (e.g., ultrasound device), the “output” of stand-alone software is limited to information.
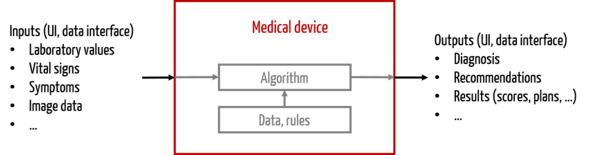
Software thus differs from other medical devices in terms of interfaces:
- User-product interface, often graphical user interface (GUI)
- Product-product interface, especially data interfaces
Therefore, for software, clinical evaluation must consider whether the information provides value to the user, for example in diagnosis or therapy selection, or whether it provides useful information to other products.
This concept will certainly sound familiar to IVD manufacturers. Thus, the benefit of an IVD always lies in the provision of appropriate medical information (see IVDR, recital 64). Clinical evidence is provided for in vitro diagnostic software through performance evaluation.
Caution:
Here, we are not referring to cases in which the software is used to control medical devices. This is because in most cases the software would be an accessory to the product in question, for which a clinical evaluation should already exist.
b) Special benefit of stand-alone software
Stand-alone software serves one of the following purposes:
- Diagnosis or provision of diagnosis-relevant information (medical devices/IVD)
- Selection of appropriate therapies, e.g., selection of an appropriate drug or decision on chemotherapy or radiation (medical devices/IVD)
- Performing the therapy itself, e.g., calculating the insertion angle of a surgical instrument, radiation planning, dose calculation (medical devices only)
Additional information
The Guidance Document MDCG 2019-11 “Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR” provides guidance on distinguishing software as a medical device or IVD.
c) Clinical evaluation or performance evaluation based on verification and validation
Clinical evaluators and performance assessors often question what aspects of software products they need to evaluate in the first place. Most of the time it makes sense to focus on the algorithms, e.g.:
- Does an image processing algorithm detect tumors with sufficient probability?
- Does the algorithm of a drug therapy safety system help to identify contraindications and interactions and to avoid medication errors?
Sometimes, these algorithms are very mundane. The question then arises as to what clinical evaluation or performance evaluation of software should yield in terms of insights. In some cases, it may be sufficient to argue on the basis of verification results.
If, on the other hand, the algorithms are more complex, proof is needed that they deliver the promised benefit, i.e., that they are clinically valid. This is precisely the approach taken in the MDCG document MDCG 2020-1.
3. Three pieces of evidence for the clinical evaluation of software according to MDCG 2020-1
According to MDCG, the manufacturer must provide three pieces of evidence:
- Scientific validity
- Technical performance/analytical performance
- Clinical performance
In providing evidence, the manufacturer should go through five different phases:
- Planning
- Data collection
- Data evaluation
- Data analysis
- Documentation
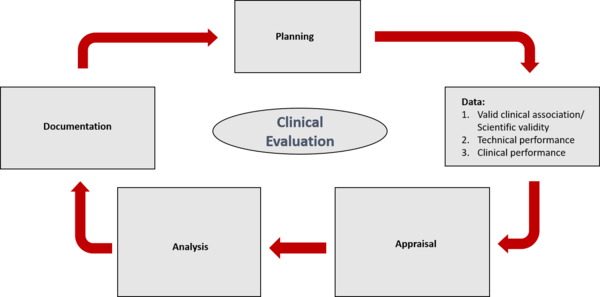
These three types of evidence are presented below.
a) Proof 1: Scientific validity
Source: MDCG 2020-1
Definition: VALID CLINICAL ASSOCIATION/SCIENTIFIC VALIDITY
“... is understood as the extent to which the MDSW’s output (e.g., concept, conclusion, calculations), based on the inputs and algorithms selected, is associated with the targeted physiological state or clinical condition. This association should be well founded or clinically accepted”
MDCG 2020-1
Scientific validity, then, is the evidence that the “outputs” of the product (in this case, the software) are associated with a clinical or physiological condition.
One example is the calculation of a score, which must actually be a measure of a certain risk.
To demonstrate scientific evidence, manufacturers should use the following sources:
- Scientific literature (peer-reviewed articles, conference papers)
- Recommendations by the authorities
- Scientific databases, including those with results from clinical studies
- Expert opinions, e.g., in books and guidelines
b) Proof 2: Technical performance
Definition: TECHNICAL PERFORMANCE/ANALYTICAL PERFORMANCE
“... is the demonstration of the MDSW’s ability to accurately, reliably, and precisely generate the intended output from the input data”
MDCG 2020-1
Verification of technical performance is thus the demonstration of the software product’s ability to accurately, reliably, and precisely produce the intended output from the input data.
Manufacturers prove technical performance in the context of verification and validation (V&V). In doing so, they should draw on the following, as appropriate:
- 1. Verification activities, e.g.:
- Unit tests
- Integration tests
- System tests
- 2. Validation activities, e.g. based on:
- Curated databases or registers
- Reference databases
- Previously collected patient data
Typically, medical device manufacturers reference these V&V results when clinically evaluating software.
IVD manufacturers finally evaluate the technical performance of the software during the analytical performance evaluation of the IVD and take into account the available V&V results.
c) Proof 3: Clinical performance
Definition: CLINICAL PERFORMANCE
“... is the demonstration of an MDSW’s ability to yield clinically relevant output in accordance with the intended purpose”
MDCG 2020-1
With regard to clinical performance, the manufacturer must demonstrate clinical relevance to the intended purpose of the device. The clinical relevance of medical device software is shown to have a positive impact on
- an individual’s health, expressed in terms of measurable, patient-relevant clinical endpoints, including endpoint(s) related to diagnosis, prediction of risk, prediction of treatment response(s); or
- aspects related to its function, such as those of screening, monitoring, diagnosing, or helping to diagnose patients; or
- patient management or public health.
These data may come from clinical studies conducted by the manufacturer as well as from other sources. Manufacturers are also encouraged to use existing data from alternative processes and products.
For example, you should refer to the following information to assess clinical performance:
- Clinical/diagnostic sensitivity and/or clinical/diagnostic specificity
- Positive/negative predictive value
- Number needed to treat (number of patients who need to be treated for a disease or preventively to avoid an additional event such as (secondary) illness or death)
- Number needed to harm (number of patients that need to be diagnosed/treated to have an adverse effect on a patient)
- Ease of use/user interface
- Confidence interval(s)
d) Summary of steps for clinical evaluation of software
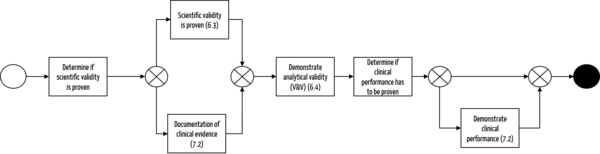
The following flowchart once again shows the main steps in the preparation of the clinical evaluation:
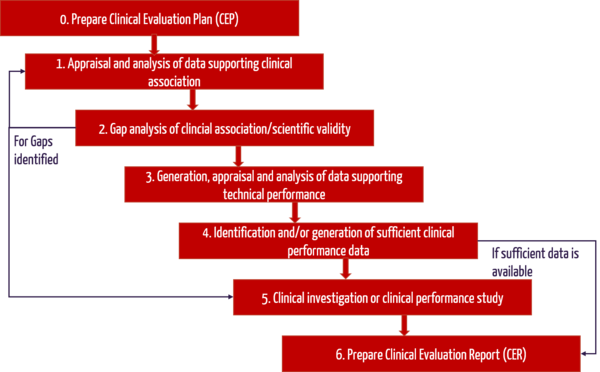
4. Clinical evaluation of software after it has been placed on the market (post-market)
Manufacturers must actively and continuously monitor the safety, effectiveness, and performance of the MDSW after it is placed on the market.
As with all manufacturers, information may include, but is not limited to, complaints, direct end-user feedback, or newly published research/guidelines. Of particular relevance to MDSW, however, is the collection of REAL-WORLD PERFORMANCE DATA, as this is relatively easy to obtain with software.
This allows the very quick and effective
- timely detection and correction of malfunctions,
- detection of systematic abuse,
- understanding of user interactions and
ongoing monitoring of intended clinical performance (e.g., sensitivity, specificity).
Additional information
Here you will find information on Post-Market Surveillance and the Post-Market Surveillance Plan.
5. Assessment of the level of evidence and data
First and foremost, manufacturers must decide for themselves whether the level of data is sufficient for each step and justify this if necessary. This involves both the quantity and the quality of the data. This evaluation can be guided by the following questions:
a) Sufficient quantity
- Do the data support the intended use, indications, target populations, clinical claims, and contraindications?
- Have the clinical risks and analytical performance or clinical performance been investigated?
- Have relevant MDSW characteristics such as data input and output, algorithms used, or type of interconnection been considered in generating the data to support the performance of the product?
- What is the level of innovation or history in the market (how extensive is the body of scientific knowledge)?
b) Sufficient quality
- Was the type and design of the study or test appropriate to meet the endpoints?
- Was the data set appropriate and up to date (state of the art)?
- Was the statistical approach adequate to reach a valid conclusion?
- Have all ethical, legal, and regulatory considerations or requirements been taken into account?
- Is there any conflict of interest?
c) Demonstration of compliance without clinical data
In some cases, particularly for lower risk class I and IIa medical device software, clinical data is often not adequate to demonstrate compliance. Both the MDR (Article 61, Section 10) and MDCG document 2020-1 allow an exception.
Demonstration of compliance with essential safety and performance requirements can be based solely on the results of non-clinical test methods including performance evaluation, technical testing (“bench testing”) and pre-clinical evaluation. However, this must be justified in any case. The following points must be taken into account:
- Manufacturer’s risk management
- Consideration of the special characteristics of the interaction between the product and the human body
Intended clinical performance and manufacturer’s performance data
Caution:
NOTE: Manufacturers must still prepare a clinical evaluation that includes, at a minimum, the rationale for the exemption pathway, evidence of the nonclinical test methods, and a statement on the state of the art.
If certain performance requirements are not applicable for in vitro diagnostic devices, they can be justifiably excluded. IVD manufacturers describe their intended performance evaluation strategy in the product-specific performance evaluation plan. There they also set out the state of the art. In the performance evaluation report, the results on the clinical evidence and the benefit-risk ratio are finally summarized and evaluated.
6. FDA & IMDRF on Clinical Evaluation of Software as a Medical Device (SaMD)
Together with the IMDRF, the FDA has already published a very similar Guidance document “Software as a Medical Device: Clinical Evaluation” published. It requires that manufacturers verify the clinical validity of the product with the clinical evaluation of medical device software or the performance evaluation of IVD software. In doing so, they demonstrate that the intended purpose is fulfilled and that there are no unacceptable risks. Some of the definitions differ from those of the MDCG. This may lead to a lot of extra work if both countries have to be served.
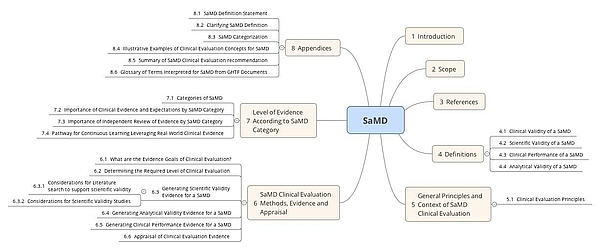
The FDA provides guidance on what steps need to be taken depending on certain parameters (risk class, degree of innovation, etc.).
a) Determine class for the software
FDA uses a classification from other IMDRF documents for clinical evaluation of software:
| Significance of information provided by SaMD to healthcare decision | ||
State of healthcare
| Treat or diagnose | Drive clinical management | Inform clinical management |
Critical | IV.i | III.i | II.i |
Serious | III.ii | II.ii | I.ii |
Non-serious | II.iii | I.iii | I.i |
As examples of the areas of application, the FDA cites:
- “Treat or diagnose”
- Treat
- Provide therapy to a human body using other means
- Diagnose
- Detect
- Screen
- Prevent
- Mitigate
- Lead to an immediate or near-term action
- “Drive clinical management”
- Aid in treatment
- Provide enhanced support to safe and effective use of medicinal products
- Aid in diagnosis
- Help predict risk of a disease or condition
- Aid to making a definitive diagnosis
- Triage early signs of a disease or condition
- Identify early signs of a disease or condition.
- “Inform clinical management”
- Inform of options for treatment
- Inform of options for diagnosis
- Inform of options for prevention
- Aggregate relevant clinical information
- Will not trigger an immediate or near-term action
b) Demonstrate evidence depending on the class
Depending on the class, manufacturers must demonstrate the following:
| Green class | Yellow class | Red class |
Analytical validity | Yes | Yes | Yes |
Scientific validity | Yes | Yes | Yes
|
Clinical performance | Only for new processes | Only for diagnostic devices of class II.iii and for new procedures | Only for diagnostic products and for new procedures |
Independent review (objective and competent person outside the manufacturer) | No | No | Yes |
c) Determine necessary evidence
If the manufacturer has competence and capacity, the next step is to establish the necessary evidence. In other words, the manufacturer should use the above table to decide what evidence to provide:
- Analytical validity
- Scientific validity
- Clinical performance
What lies behind these three aspects is explained above.
d) Collect data
The data can be collected as described above. Common data sources at FDA also include:
- Scientific literature
- Results of the verification and validation of the software
- Existing post-market data for own and/or third-party equivalent products
- Clinical trial results
e) Evaluate data and prepare clinical evaluation of software
At the FDA, the next step is to evaluate the data and write the clinical evaluation or (for IVDs) the final performance evaluation report. For medical devices the MEDDEV 2.7/1 provides guidance on the structure of clinical evaluations, even though the MEDDEV document does not have the FDA in scope.
Additional information
The Audit Guarantor includes clinical assessment videos as well as clinical assessment templates with examples and instructions for completion.
f) Conclusion on the guidance documents from IMDRF or FDA
The clinical evaluation or performance evaluation effort should be adjusted based on risk. The FDA proposes proceeding as follows in the clinical evaluation of software:
The IMDRF’s text is not easy to digest. The authors do a good job of presenting the various aspects of clinical evaluation or performance evaluation of software. They refer to many other publications by the same organization.
7. Summary
In the document MDCG 2020-1 on clinical evaluation or performance evaluation of software, the European Medical Device Coordination Group attempts to take account of the special requirements for software products. The authors present the various aspects of the clinical evaluation of software as a medical device or IVD quite well. The approach is much more pragmatic than the previous one.
However, some things still remain vague. Some concrete examples in the appendix help to clarify what is meant, as well as when which step of the algorithm is obligatory and when it is not. Here, a more precise demarcation of the requirements (e.g., based on the classification of the functionalities or classification of the software) would be very desirable.
It also lacks any reference to other MDCG guidance documents regarding general aspects of clinical evaluation, e.g., requirements for authors.
The FDA also offers a guidance document that takes a very similar approach. This shows much more clearly when which proof characteristics are required.
As with any clinical evaluation, the requirements of the MDCG guidance document cannot be implemented without a scientifically and medically competent team.
The Johner Institute specializes in preparing clinical evaluations for medical devices for compliance with MEDDEV 2.7/1 and MDCG 2020-1 and ensuring that they are accepted by notified bodies.
The Johner Institute’s IVD team will also be happy to assist you in developing a performance evaluation strategy for your IVD software.
Please let us know (e.g., via the Contact form) if you are interested in support from the experts of the Johner Institute for clinical evaluations or performance evaluations.

Author:
Andrea Seeck

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.