ASCA: Accreditation Scheme for Conformity Assessment
Expert: Mario Klessascheck
ASCA stands for Accreditation Scheme for Conformity Assessment. The procedure is intended to accelerate conformity assessments and, thus, approval procedures. However, it is not applicable to all devices or all markets.
This article explains who benefits from ASCA and how the procedure works.
1. ASCA – The basics
a) Who is affected by ASCA?
ASCA primarily affects these stakeholders:
1. Medical device manufacturers whose devices fall under the IEC 60601, IEC 80601, IEC 61010, and ISO 10993 families of standards
2. Test houses, which test the devices for conformity with these standards
3. Accreditors (Accreditation Bodies) of the test houses
4. Regulatory authorities, in particular the FDA
Currently, the procedure is limited to the following approval processes in the U.S.: 510(k), De Novo, Premarket Approval (PMA), and Investigational Device Exemption (IDE).
b) What is ASCA?
ASCA is a voluntary pilot program for a new accreditation system in which
- testing laboratories undergo additional qualification/accreditation and
- test reports are standardized.
The FDA trusts the test methods and outputs (primarily the ASCA Summary Test Report) of ASCA-accredited test houses and test laboratories. It does not intend to request additional information; this will expedite approval. The procedure is described below.
c) Why is ASCA needed?
The FDA criticizes the occurrence of too many incidents with medical devices. In addition to the manufacturers, the FDA has identified the inadequate quality of the test houses as one reason for this.
The authority has also recognized that medical devices are becoming increasingly complex. On the one hand, this is overtaxing the agency's own inspectors, and on the other, it is leading to more and more queries by inspectors from experts and manufacturers.
All this ties up unnecessary resources at the FDA and delays the approval of medical devices.
d) What are the benefits of ASCA?
Thus, the ASCA program is intended to achieve the following objectives:
- Increase patient safety
- Improve patient care through more readily available medical devices
- Reduce efforts of the authority and other stakeholders (e.g., manufacturers)
- Expedite approval processes
Probably the highest benefit is for FDA investigators. They must read less and consult less frequently with internal and external experts.
ASCA-accredited testing laboratories are likely to benefit from increasing numbers of orders.
Whether the faster approval times outweigh the higher costs for manufacturers depends on the individual case.
2. Procedure
a) Qualification of the test houses
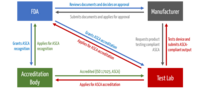
The test houses must be accredited by an accreditation body. Then they can apply to the FDA for the ASCA program. Only then are they allowed to test devices that benefit from accelerated approval (see Fig. 1).
b) Approval of the devices
The manufacturers develop their devices as usual. For regulatory approval, they proceed as follows:
1. Plan regulatory strategy (e.g., select and sequence target markets).
2. Select a list of standards with which conformity is to be declared (This also includes standards for which ASCA should not or cannot be applied).
3. Select a test laboratory (A list can be found here; in Germany, there is currently one accredited test laboratory).
4. Prepare a suitable test plan with the test laboratory (this includes the necessary and applicable performance tests, which must also be performed by the test laboratory, as well as the test conditions, additions or changes to tests, and acceptance criteria according to risk acceptance).
5. Test the device and summarize test results in the ASCA summary test report.
6. Compile submission documents to FDA (This includes a "cover letter" with reference to ASCA tests, Summary Test Report, ASCA Declaration of conformity, and other documents).
7. Submit documents to the FDA (already, when applying via eStar, one can select that the inspection will be carried out according to ASCA, see Fig. 2).

It is then the FDA's task to review the dossier. During the approval process, the device goes through the usual phases (see Fig. 3).
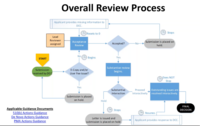
The actual time-saving concerns the steps "Substantive Interaction" and "Outstanding issues are resolved interactively." The program should shorten these, and the iterations should even be avoided if possible.
Further information
The FDA provides extensive information on its website. Particularly relevant are the following:
3. Requirements for the ASCA test laboratories
a) ISO/IEC 17025
The QM standard ISO/IEC 17025 applies to test laboratories, and conformity with this standard is a prerequisite. ASCA specifies the requirements of this standard.
Example
With regard to training and competence of personnel, ASCA-accredited test houses must meet the following requirements:
- Technical personnel must understand the requirements of the standards (e.g., IEC 60601-1). In particular, this includes underlying concepts such as basic safety and essential performance.
- The laboratory is required to implement a program of education and training for technical personnel.
- This program obligates training and documentation of the training and its outputs.
- It must include ongoing training (on-the-job or classroom training) that occurs at specified intervals or/and when test standards or methods are updated or newly developed.
- The testing laboratory must already specify the requirement profiles precisely in the job descriptions.
b) Overview of ASCA requirements
The requirements concern not only the competence of the personnel but also the following:
- Equipment of the testing laboratory
- Verification and validation methods
- Preparation and guidance of test plans and test instructions
- Consideration of the intended purpose and the intended use of the device in the test plans
- Preparation of test reports, especially the ASCA summary test report
- Scope and content of test reports (for example, the testing laboratory must communicate all opinions and interpretations to the manufacturers in writing, including concerns about basic safety and essential performance)
c) Additional FDA requirements
The FDA also specifies requirements for regulatory documents, specifically:
- Cover Letter
- Declaration of Conformity
- ASCA Summary Test Report
- Test plan and procedure
- Acceptance criteria and outputs justifying the safety claim
4. Tips and hints
a) Have the device pre-tested
The ASCA Summary Test Report becomes a central document that significantly reduces testing time. It is a prerequisite that the report is complete and accurate. The Summary Test Report will also mention concerns that the test house has and mention if a test had to be repeated or if changes to the device were necessary.
Therefore, it may be useful to also have the device tested during development to avoid surprises during the ASCA inspection.
b) Create precise safety concept
Frequently encountered difficulties include insufficiently precise or even missing documentation of the safety concepts or the system architecture.
A blanket statement such as "The device must meet the requirements of IEC 60601-1" will no longer suffice. Notified bodies in Europe are already taking a closer look today, and testing laboratories must do the same.
This means that manufacturers should specify the safety requirements and safety goals regarding single-fault safety during the design phase and then benefit from the safety concept as a guideline to build safe systems. The basic standard for functional safety IEC 61508 can provide informative assistance.
c) Specify acceptance criteria
Test plans should specify appropriate test methods for performance testing with accurate acceptance criteria. Both manufacturers and test laboratories should note that systems have dynamic behavior. This is often not tested.
Test labs are then able to create an appropriate test plan more quickly and define an appropriate test setup with the manufacturer.
Other important information for the test laboratory is:
- Intended purpose/intended use, including intended use environment and intended user
- Comprehensive product description
- Systematic derivation of essential performance characteristics
- Discussion of the special properties of the technologies used
- Information on labeling, especially with regard to the intended purpose and use under various circumstances
- Evidence of performance testing, such as test reports and clinical data
- Derivation of limit values and acceptance criteria
- Description of any changes to tests or additional tests that are necessary
This information can either be included in the product description, in the architecture document, or in individual documents. There is no default for the structure. However, this is not new, because notified bodies and the FDA already require all this information anyway.
d) Consider costs
The FDA estimates the additional effort of the test laboratories to prepare the reports to be about 47 hours, i.e., about one working week. This is likely to entail costs in the order of EUR 10,000.
5. Summary and conclusion
a) Possible disadvantages
Test laboratories have a higher overhead, which drives up costs and may prevent smaller test laboratories from participating in the program. This could result in limited testing capacity.
The test labs will pass the cost on to the manufacturers. One possible consequence is that companies that cannot afford the cost will not be able to benefit from FDA's expedited procedures. If so, the FDA will not achieve its objective of getting innovative medical devices to market faster.
b) Possible advantages
Precise requirements also have advantages:
- They save all parties involved (manufacturers, test houses, approval bodies) unnecessary discussions.
- Manufacturers know early on what to expect. Accordingly, they can ensure conformity at an early stage, which saves expensive rework and iterations.
- This makes testing and approval procedures faster and easier to plan.
c) Outlook
The FDA plans to convert the ASCA pilot program into a permanent program. By 2024, the ASCA team will assess whether the objectives have been met.
It is anticipated that the ASCA procedures and the CB procedure will be combined. This will allow regulatory bodies other than the FDA to accept the tests and test results, at least somewhat fulfilling the desire to harmonize regulatory requirements worldwide.
Support
Benefit from the Johner Institute's help to pass all approval procedures quickly and with minimal effort, with precise documentation and safe medical devices.
We support you in preparing your documents (e.g., safety concept, risk analysis, defining essential performance characteristics, test plans, test strategy) or taking over this work entirely.
Our experts also assist you in reviewing all documents, such as the architecture (compliant with ISH1, with functional safety methods) and all other "submission documents."

Author:
Mario Klessascheck

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.