Software Safety Classes (IEC 62304) versus Levels of Concern (FDA)
Both, European and US regulations, distinguish three different categories of medical device software, the software safety classes accordingly to IEC 62304 respectively the FDA levels of concern. Frequently manufactures confuse both.
Software safety classes
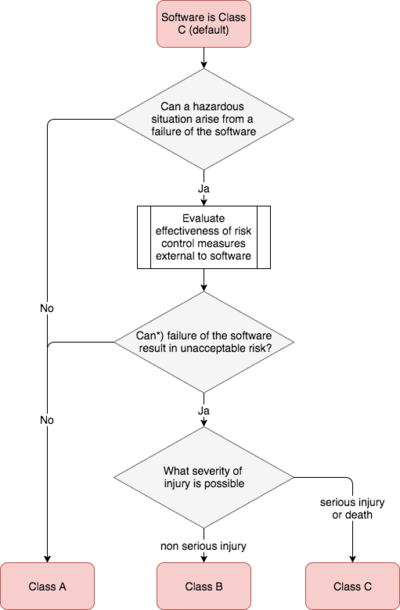
IEC 62304:2006 + Amendment 2015 define the software safety classes as follows:
Definition: Software Safety Class
„The SOFTWARE SYSTEM is software safety class A if:
- the SOFTWARE SYSTEM cannot contribute to a HAZARDOUS SITUATION; or
- the SOFTWARE SYSTEM can contribute to a HAZARDOUS SITUATION which does not result in unacceptable RISK after consideration of RISK CONTROL measures external to the SOFTWARE SYSTEM.
The SOFTWARE SYSTEM is software safety class B if:
- the SOFTWARE SYSTEM can contribute to a HAZARDOUS SITUATION which results in unacceptable RISK after consideration of RISK CONTROL measures external to the SOFTWARE SYSTEM and the resulting possible HARM is non-SERIOUS INJURY.
The SOFTWARE SYSTEM is software safety class C if:
- the SOFTWARE SYSTEM can contribute to a HAZARDOUS SITUATION which results in unacceptable RISK after consideration of RISK CONTROL measures external to the SOFTWARE SYSTEM and the resulting possible HARM is death or SERIOUS INJURY“
IEC 62304:2006 + A1:2015
1. Definition of safety classes and levels of concern
The following diagram summarizes this classification:
Level of Concern
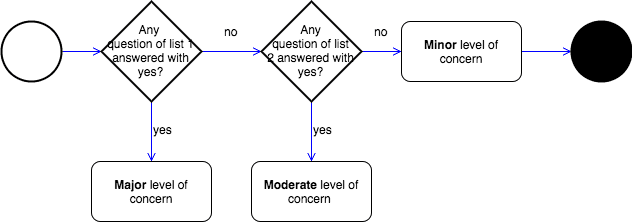
The FDA defines the Levels of Concern as follows:
Definition: Level of Concern
- Minor: We believe the level of concern is Minor if failures or latent design flaws are unlikely to cause any injury to the patient or operator.
- Moderate: We believe the level of concern is Moderate if a failure or latent design flaw could directly result in minor injury to the patient or operator. […].
- Major: We believe the level of concern is Major if a failure or latent flaw could directly result in death or serious injury to the patient or operator. […]
FDA guidance document: Content of the premarket submission for software contained in medical devices
To determine the classification, FDA defines a set of lists. If a question is answered the software falls in the respective class. Examples of questions in list 1 are:
- Is the Software Device intended to be used in combination with a drug or biologic?
- Is the Software Device an accessory to a medical device that has a Major Level of Concern?
- Prior to mitigation of hazards, could a failure of the Software Device result in death or serious injury, either to a patient or to a user of the device? E.g. Does the Software Device control a life supporting or life sustaining function?
Please note that both definitions primarily are dependent on the severity of potential harms and not on risks. I.e. the probability of this harms is not what determines the software safety class respectively the level of concern.
2. Goals of classification
The IEC 62304 introduces the software safety classes to determine the extent of documentation to be complied.
| 5.1 | 5.2 | 5.3 | 5.4 | 5.5 | 5.6 | 5.7 | 5.8 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | x | x | -- | -- | -- | -- | x | x | x | x | x |
| B | x | x | x | -- | (x) | x | x | x | x | x | x |
| C | x | x | x | x | x | x | x | x | x | x | x |
Table 1: The documentation depends on the safety class IEC 62304. E.g. for class A software no software architecture (chapter 5.3) is required. The numbers correspond to the chapters of the standard.
The FDA uses the levels of concern to determine the amount of documentation to be submitted. I.e. the level of concern does not influence the documentation to be compiled.
| Software documentation | Minor | Moderate | Major |
|---|---|---|---|
| Level of concern | x | x | x |
| Software description | x | x | x |
| Device hazard analysis | x | x | x |
| Software requirements specification | (x) | x | x |
| Architecture design chart | x | x | |
| Software design specification | x | x | |
| Traceability analysis | x | x | x |
| Description of software environment | x | x | |
| Verification and Validation documentation | (x) | x | x |
| Revision level history | x | x | x |
| Unresolved anomalies | x | x |
Warning!
If you market your products in the US, the software safety class is irrelevant. You have to document according to class C anyhow. Do not confuse compilation and submission of documents!
3. Reduction of software safety class respectively level of concern
Reduction of software safety class
IEC 62304 permits a reduction of the software safety class by means that are external to the software only. Examples are:
- Physical hardware e.g. a stopper
- Other component containing hardware (electronics) and even software e.g. a watchdog
- User e.g. responding to a warning (not in IfU) or pressing an emergency stop
Reduction of level of concern
The FDA expects measures to mitigate risks. However, the levels of concern are determined prior to these measures. I.e. a reduction of the level of concern does not impact the documentation to be submitted.
4. Changes: end of the “level of concern”
On November 4, 2021, the FDA published the draft of a guidance document entitled Content of Premarket Submissions for Device Software Functions. This document is intended to replace the guidance document that introduced the level of concern.
This new guidance document only differentiates between two classes.
a) Defining the classes
The FDA no longer defines three “levels of concern.” Instead, it now defines two “documentation levels”:
- Basic Documentation Level
- Enhanced Documentation Level
Software requires “Enhanced Documentation" if at least one of the following conditions is met:
This device that contains the software or is the software:
- Is a combination product
- Is intended for use in transfusion medicine or organ donation (determination of organ donor and recipient compatibility)
- Is classified as class III
- Can lead to death or serious injury in the event of a fault
With regard to the fourth point, the FDA refers to reasonably foreseeable risks.
b) Consequence of classification
Like the level of concern, the classification also affects the scope of the (software) documentation to be submitted:
Element of software documentation | Basic Documentation | Enhanced Documentation |
Determination of the documentation class | x | x |
Software description | x | x |
System and software architecture | x | x |
Risk Management | x | x |
Software requirements specification | x | x |
Software design specification | — | x |
Documentation of software development and maintenance | x | x (expanded) |
Software testing | x (without details of unit and integration testing) | x |
Version history | x | x |
List and evaluation of anomalies | x | x |
The new requirements are very similar to the previous moderate and major levels. This means that manufacturers cannot submit software documentation that only meets the very lax requirements of the minor level of concern.
c) Evaluation
The FDA makes extensive references to IEC 62304. However, from the publication of the new version of the guidance document at the latest, a procedure as per class A is no longer permitted. In addition, manufacturers should be aware that the amount of documentation to be submitted is independent of the amount of documentation to be created.
The Johner Institute recommends that manufacturers work and create documentation in conformity with the requirements of safety class C.

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.