Software Specifics
Software risk management are either the risk management activities that medical device manufacturers need carry out for standalone software or the part of the risk management activities that are necessary in order to identify hazards arising from the use of embedded software.
Very often manufacturers are not able to fulfil the regulatory requirements of software risk management. This article shall give you valuable tips for lean software risk management that can help you avoid unnecessary effort and achieve compliance
Regulatory requirements for the software risk management
The EU regulation for medical devices (MDR) doesn’t differentiate between devices with or without software, with regards to risk management. The ISO 14971, the harmonized risk management standard, has no explicit software risk management. Only the IEC 62304 has an explicit chapter “software risk management process”. But the IEC 62304 with its philosophy, that errors in software are to be assumed to occur with 100% probability, seems to disagree with the principle of the ISO 14971 and its definition of risk, namely the combination of severity and likelihood of damage.
IEC 62304 sets out a few software-specific requirements with respect to software risk management. These include the monitoring of SOUP components.
Particularities of software risk management
Software alone does not lead to harm. Software can not hurt or even kill. At least not directly. The damage is always done indirectly via physical devices (e.g. contusion by a table, beaming device) or for example by people who treat patients incorrectly or not all. This makes software risk management so challenging.
So, if you want to carry out software risk management, then you need to analyse the cause and effect chain from the defective software to the patient (or the user).
Unlike physical devices the hazard arising from standalone software is not on the outer border of the product (the display for standalone software). For physical devices, the electrical energy, mechanical energy, radiation energy or the chemical materials (all hazards) are always on the outer border of the medical device. According to the definition of hazard – a potential source of harm - the wrong message display can be seen as a hazard.
The ISO 14971:2012 (unfortunately) spoke about informational hazards, but these are causes rather than hazards, at least as long as the instruction manual doesn't fall on your foot.
The new version of ISO 14971:2019 speaks of "performance-related hazards" and is thus closer to the MDR requirement for elimination/reduction of risks and performance impairments (Annex I 17.1). Performance-related hazards refer, for example, to the availability of relevant data, the absence of alarms, or the incorrect update rate of monitoring data in a monitoring system.
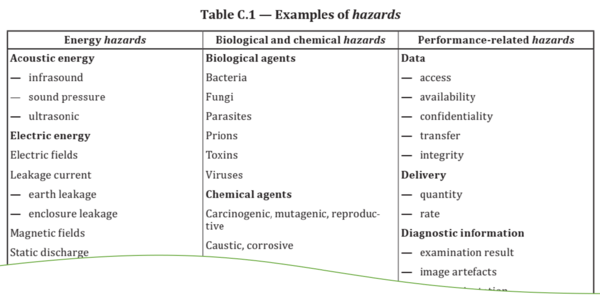
We recommend to at least consider refining the definition: Not every element in the chain of causes is a hazard, but only the last element before the hazardous situation.
If this supplemental definition is adopted, it is no longer the wrong medication information in the display of the software but the wrong drug that is the hazard - a chemical hazard.
This refined definition often also helps in the use of the term "hazardous situation," the circumstances in which people, property, or the environment are exposed to one or more hazards: A patient may never be exposed to the wrong information on a display because only the physician or practitioner sees it. But the patient is exposed to the wrong drug.
Make sure that you do not confuse causes, hazards, hazardous situations, risks and damages in risk management and stay consistent. Your Risk Management file is otherwise going to be a problem during the audit. Let us know if we can help (Contact via web form).
Tips for Software Risk Management
a) Right granularity
"How deep does the risk analysis of the software components need to go within the architecture? Do you have to go down to class level or even down to the method level? " These are common questions raised in the “micro-consulting of Johner Institute.
A rule defining how “deep you must go” cannot be given. Start with a separation between visible system misbehavior of a product and the resulting risks and the cause and effect chain leading to this external system misbehavior (bottom-up). This corresponds to an FMEA (failure mode and effect analysis).
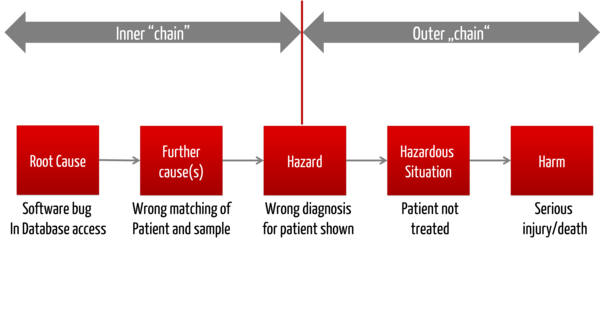
Only in the case of a risk following a given visible external system misbehavior, you have to investigate the cause and effect chain "in the device", i.e. investigate down to component level. This corresponds to an FTA (fault tree analysis) from the outer border of the device in the direction of the components (top-down).
This second analysis has the following objectives:
- You want to find out the causes and possibly the probabilities for this misbehavior.
Note: These causes (e.g. faulty components) have a possibility of misbehaviour, but no severity. This only exists in the outer chain. - You want to investigate whether there are risk control measures, in order to precisely interrupt this chain of causes.
You can end with the analysis, once you have defined a risk control measure that cuts the fault tree. The higher you can cut the tree, the better it is (ideally not within the same software system).
Even if I don’t appreciate answers like "it depends", it is still the only possible answer to the question of the depth of the analysis. Most companies examine too deeply but not broad enough. This leads to excessive risk tables that cause a lot of work, but do not provide real insights. Many risks remain undetected.
Just take a look at our e-learning platform, where you can access a number of video training sessions on risk management according to ISO 14971 in software.
b) Composition of the team for the software risk management
Risk management is a team sport in which should participate:
- Risk Manager: expert regarding regulations, methods, documentation.
- Software or system architect: Knows the internal cause and effect chain and can (only) estimate probabilities that the software or the system will behave incorrectly.
- Context Expert: can assess the probability that an external malfunction of the system results in a hazardous situation.
- Doctors: can judge with what probability a hazardous situation leads to a harm of a certain severity.
So, you should not leave the software risk management to the software developers alone. But I refer only to the roles, not specific individuals who may hold multiple roles.
Risk management for software and software safety classes
You may classify a software system after an (initial) risk analysis: It may not be absolutely necessary to consider the probabilities exactly, but definitely the severity of harm. If the result of this analysis shows that no harm can be caused by the software, then the software safety class A is justified. Accordingly, you don’t have to look closer at the software, i.e. document the software in much detail. However, keep in mind that software development according to class A is not really state of the art.
Risk management has to be started before the safety classification, but it doesn’t end with. For example, you have to monitor, as part of chage manmagement and post market surveillance that the safety class A, was/is still correct.
In other words, you must always comply with both standards ISO 14971 and IEC 62304. Or to be more exact: You must meet the basic safety and performance requirements of the MDR with respect to risk management and software lifecycle.
If one has an entirely uncritical (stand-alone) software, then the amount of documentation is automatically reduced to a minimum. Ultimately, one only needs to cleanly prove that it really is uncritical.
For standalone software of safety class A, you might ask yourself if this is really a medical device ...

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.