The FDA's Breakthrough Devices Program

The FDA is trying to facilitate quicker access for critically ill patients to novel medical devices through a new program for breakthrough devices.
In December 2018, it published a guidance document on the program.
In this article, we will explain how the Breakthrough Devices Program works and what requirements manufacturers must meet in order to be able to participate in it.
Aims of the FDA's Breakthrough Devices Program
The FDA's aim is to open up a pathway for manufacturers to get their medical devices on the market faster. However, the program is only aimed at novel devices that help patients who are suffering a life-threatening disease or who are suffering an irreversibly debilitating disease or condition.
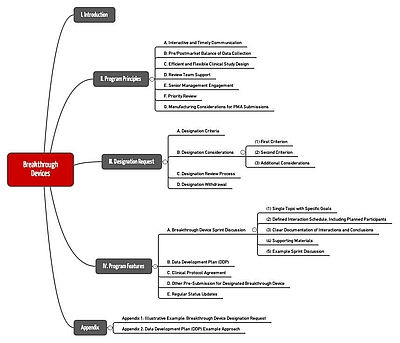
The most important aspects of the Breakthrough Devices Program
The Breakthrough Devices Program is not a new form of approval. Therefore, it is not an alternative to a Premarket Notification according to 510(k), a Premarket Approval, or a “De Novo Request”. Rather, the FDA wants this program to enable these existing approval procedures to be made quicker.
The program is open to medical devices and combination products. It replaces the following programs:
- Expedited Access Pathway
- Priority Review
- Innovation Pathway
How is the FDA speeding up the approval of breakthrough devices?
The FDA offers several improvements to the manufacturers included in this program:
- Interactive communication
Manufacturers can interact directly with the FDA, for example to discuss how clinical trials should be set up and to discuss regulatory documents. - Higher priority
The FDA will prioritize approvals (e.g. 510(k), PMAs). The same applies for Q-Submissions (for example, Premarket request). Applications for an Investigational Device Exemption (IDE), i.e. the authorization to use a devices in clinical studies, will also be prioritized. - Flexibility with regard to clinical studies
If there are no safety concerns, the FDA will allow part of the data to be collected at the beginning of the post-market phase. In view of the big (life-saving) benefits, the FDA is also prepared to accept higher risks on a temporary basis. The FDA will allow the study design to be adapted during the clinical study and the approval process. - Dedicated, available and well-trained review teams
The FDA will provide a sufficient number of well-trained staff. They will also have expertise with regard to the application of the Breakthrough Devices Program. Even higher level FDA management will be involved. - Quality management
The FDA may even be willing to turn a blind with regard to the quality management system by demanding fewer documents (“FDA may accept less quality system and manufacturing information”) or by not insisting on a preapproval inspection.
Collaboration is therefore faster and more interactive - more like an agile approach.
Requirements for participation
For a manufacturer to be able to obtain approval for their devices through the Breakthrough Devices Program, it must meet several requirements.
- The medical device provides for more effective treatment or diagnosis of life-threatening or irreversibly debilitating human diseases or conditions. Examples would include strokes, heart attacks, cancer, severe trauma and ALS.
- At least one of the following criteria must be met:
- The device is a technological breakthrough (breakthrough technology). For example, a new genetic test that would allow the treatment options to be evaluated better.
- There are no approved alternatives. According to the FDA, there is currently no device that can diagnose Parkinson's disease directly.
- It offers a significant improvement compared to approved alternatives.
The device is in the best interest of patients. This would be the case if the alternatives have major side effects or if the new device significantly improves the diagnostic options and their use.
How the process works
There are two steps in the program. In the first step, the manufacturer must submit an application in which it explains why it means the above requirements. The FDA will make a decision on the application within a maximum of 60 calendar days, and will request any additional information it requires within 30 days.
In the second phase, the FDA and the manufacturer work together in sprints. During this phase, the FDA will not just be more available to the manufacturer, it will also give higher priority to the "project" than it would receive in other approval processes.
Support
The FDA has issued a detailed guidance document that you can download here.
If you are unsure whether your product qualifies for the Breakthrough Device Program, get in touch with us. Our FDA experts in Germany and the USA can quickly assess your chances.
Conclusion
The FDA has once again shown us that it is genuinely able to weigh up benefits and risks. It has recognized that patients can be harmed just as much by refusals to grant approvals or delays to approvals for medical devices as they are by medical devices that are not sufficiently safe or effective.
It would be great if there were such a program in Europe as well. Unfortunately, with the MDR we're going in completely the opposite direction.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.