Unique Device Identification (UDI) in the EU
With the UDI system, the EU has introduced an obligation to identify and register medical devices that goes far beyond what is still required under the MDD. The Medical Device Regulation (MDR) even requires a UDI for standalone software.
Read about what you need to prepare for here.
1. Objectives of the UDI system
The EU wants to be able to track medical devices quickly and easily – from manufacturer to user. This will allow a quick reaction in the event of an incident, as
- products are easier to identify (down to the individual device, batch or software version),
- the location of the devices is traceable, thus users can be found more quickly and specifically,
- illegally marketed medical devices can be found more easily.
2. How the UDI system is supposed to work
a) Organization: Assign identification number
One or more organizations should operate a system that assigns UDIs and ensures that they are unique. These organizations are called issuing entities and must remain so for a period of at least 10 years. Currently, the following organizations are designated issuing entities under MDR Article 27(2)/IVDR Article 24(2):
- GS1
- Industry Business Communications Council (HIBCC)
- ICCBBA
- Informationsstelle für Arzneispezialitäten (IFA GmbH)
b) Manufacturers: Add a UDI to products and packaging
At this point, manufacturers obtain a unique identification number for each medical device, but also for each higher-level packaging (with the exception of containers). The MDR only waives the UDI for custom-made devices and products for clinical trials.
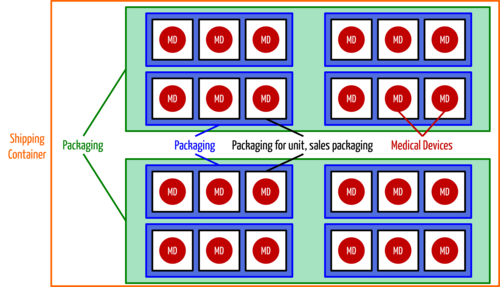
Manufacturers must include the Basic UDI in their declaration of conformity and maintain a list of all assigned UDIs as part of the technical documentation for each medical device. Further below, you will learn what the Basic UDI-DI is.
c) Operators: Save the UDIs of the medical devices
The EU encourages member states to also require operators to store the UDIs of medical devices used or purchased. The MDR obliges the health institutions to record and store the UDIs dispensed or obtained, provided the designated devices belong to class III implantable devices. This storage should preferably be done electronically (MDR, Article 27 (9)).
d) EU: Operate a UDI database
The EU must operate a database in which all UDIs are stored. It specifies which attributes per medical device must be at least taken into account. The use of this database (at least access to it) should be free of charge and public.
You can find out more about this database, the so-called EUDAMED, in our article on the European Database on Medical Devices.
e) Manufacturers: Set and update information
Manufacturers must store the Basic UDI and associated information such as product name, product version, classification (e.g. as sterile or suitable for reuse) in the database and keep it current.
3. Regulatory requirements
a) Overview
The EU formulates the requirements for the UDI system in Article 27 and in Annex VI.
Annex VI consists of several parts:
- Part A: Information to be submitted at the time of product registration, including
- manufacturer, distributor, importer
- product (UDI, type, expiration date, country where placed on the market, class and “binary” attributes such as “single use,” “contains tissue” or “contains certain substances”)
- Part B: Information to be stored in the UDI. These are partially congruent with those in Part A.
- Part C: Description of the UDI system, including
- definitions
- When which UDIs (a distinction is made between UDI-DI and UDI-PI, for example – more on this below) must be affixed where
- Human and machine readable identification
- Rules for specific product types such as implantable products, reusable products, systems, configurable products, and software.
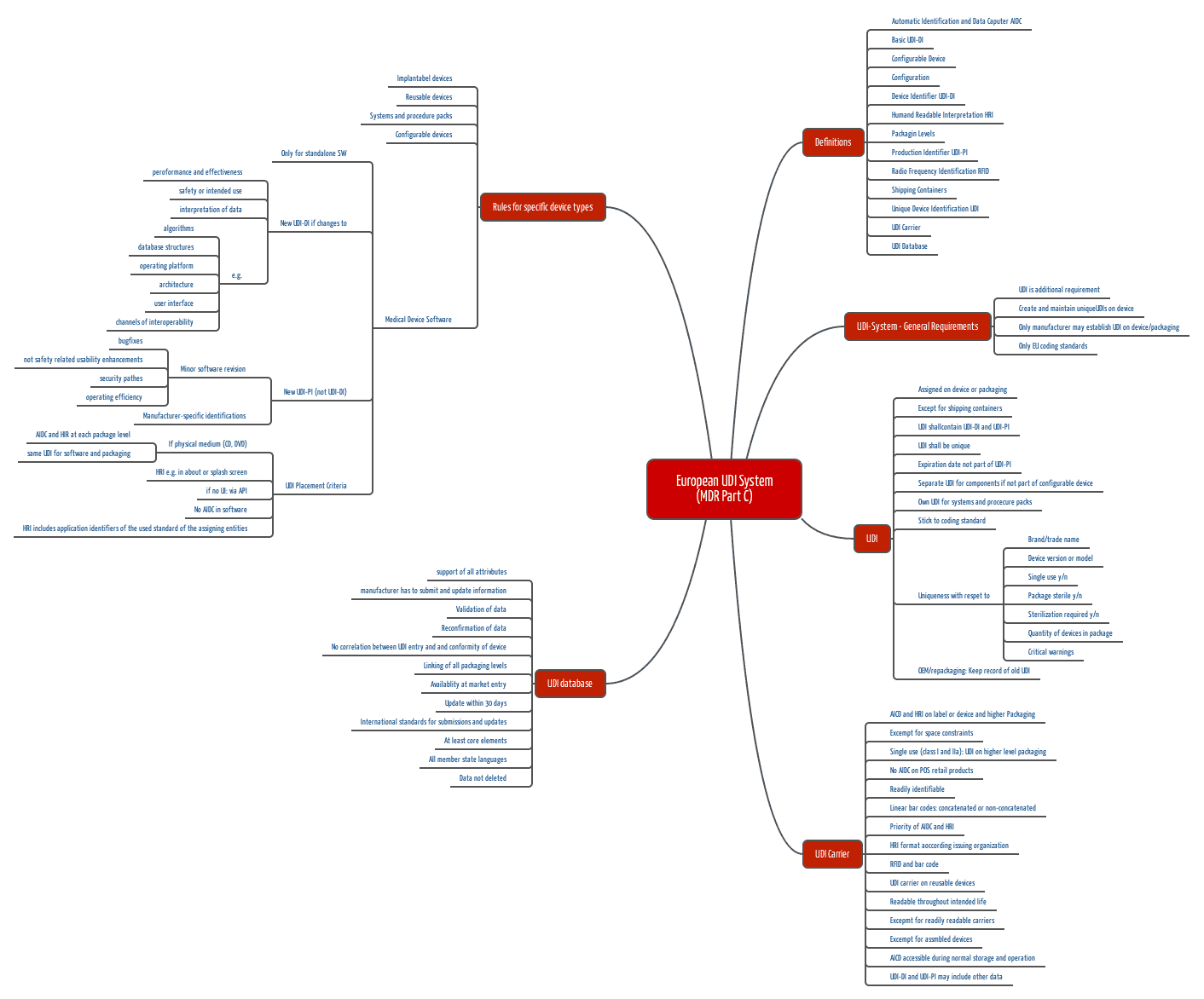
b) UDI-DI and UDI-PI
The MDR distinguishes between the two Unique Device Identifiers UDI-DI and UDI-PI:
- UDI-DI: This is the device identifier of a specific model and serves as a key in the UDI database. For example, all instances of a defibrillator of a certain type have the same UDI-DI.
- UDI-PI: The production identifier identifies each individual instance of a product or batch. Each defibrillator therefore has its own UDI-PI.
Additional information
Read more below about the difference between Basic UDI-DI and UDI-DI.
4. UDI for software
Specific rules apply to stand-alone software.
a) Allocation of a new UDI-DI
A new UDI-DI should be assigned whenever there is a change in:
- the “performance” and effectiveness
- the security
- the intended purpose
- the interpretation of data.
As examples, the MDR cites changes to:
- algorithms
- database structures
- operating system
- architecture
- user interface
- interoperability
b) Allocation of a new UDI-PI
On the other hand, “only” a new UDI-PI would be necessary for small changes such as
- bug fixes,
- security patches,
- user interface (if it only relates to usability, but not to improving security).
As a manufacturer, all changes to the third digit of the version number can be said to result in a new UDI-PI, everything else in a new UDI-DI.
Therefore, there is no requirement to allocate a unique UDI for each installation.
c) Labelling
The manufacturer must present the UDI(s):
- If there are physical media (USB, DVD, etc.), they must contain both human and machine-readable UDIs. The same applies to all outer packaging.
- The UDI must also be recognizable for users e.g. under “About” or/and the splash screen. There is no requirement to display the machine-readable UDI on the screen.
- For software without a user interface, the information must be retrievable via an API.
5. Basic UDI-DI versus UDI-DI
a) News
The EU and its Medical Device Coordination Group (MDCG) have published several guidance documents, which you will find below with comment under “News.”
b) What the Basic UDI-DI is
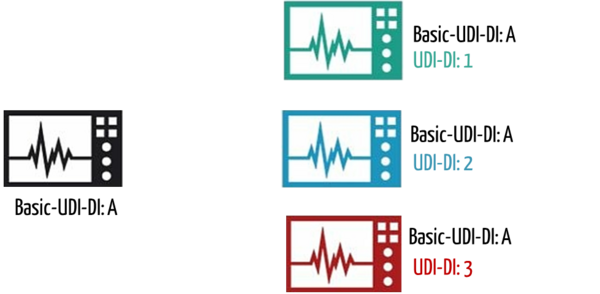
MDR and IVDR distinguish between Basic UDI-DI (“Basic UDI-DI”) and UDI-DI. The Basic UDI-DI is intended to provide a bracket for multiple variants of a medical device. Examples of such variants are:
- Products with different motor power
- Different country versions (e.g. different power supplies)
The MDR defines the UDI as follows:
The Basic UDI-DI is the primary identifier of a product model. It is the product’s identification, which is assigned at the unit of use level. It is independent of packaging units and is also the most important ordering feature for records in the UDI database.
The Basic UDI-DI must be shown in the relevant certificates and EU declarations of conformity.
Each UDI-DI belongs to exactly one Basic UDI-DI.
c) Where the Basic UDI-DI is used
The MDR and IVDR require the Basic UDI-DI to be specified in the following cases:
- On the declaration of conformity according to Article 27 (6) (MDR) / Article 24 (6) (IVDR) and Annex VI (MDR / IVDR). However, the declaration of conformity must also identify those devices for which conformity is declared under the common Basic UDI-DI.
- When registering the products in the EUDAMED according to Article 29 or Annex VI of the MDR / according to Article 26 of the IVDR.
- In the summary report on safety and clinical performance according to Article 32 of the MDR / summary report on safety and performance according to Article 29 of the IVDR.
- On the certificates of free sale according to Article 60 (MDR) / Article 55 (IVDR).
- In the technical documentation, according to Annex II (MDR/IVDR)
- In vigilance and post-market surveillance reports, e.g., MIR and PSUR (MDCG 2021-19)
- On certain EC certificates, e.g., the certificate for the assessment of technical documentation according to Annex IX of the MDR/IVDR (MDCG 2021-19)
The fastest and most cost-effective way to become compliant with UDI requirements!
Get a tool to quickly build UDI knowledge and create documents effortlessly with the medical device university. Our templates will help!
d) Which devices can be grouped under one Basic UDI-DI
According to the Guideline of the UDI Working Group, the different Basic UDI-DIs/UDI-DIs contain the following data elements:
Basic UDI-DI | UDI-DIs | UDI DIs (container package DI) |
|
|
|
Table 1: The mandatory fields are noted in bold; bold and italic are the fields that are mandatory if they are applicable. The data elements where changes result in a new Basic UDI-DI/UDI-DI are underlined.
Thus, multiple variants of a product may only display a common Basic UDI-DI if the following fields are the same:
- Single Registration Number (SRN) of the manufacturer
- Risk class
- Implantable (y/n)
- Measuring function (y/n)
- Reusable surgical element (y/n)
- Active medical device (y/n)
- Intended to supply or remove medicines (y/n)
- Medical Device Nomenclature Code (MDNC) (now EMDN Code)
However, this does not mean that manufacturers are permitted to attach a common Basic UDI-DI to all products with the same data elements specified above. According to MDCG 2018-1 v4, products may only have a common Basic UDI-DI if
- they have the same intended purpose,
- if their design is essentially the same, and
- if their manufacture, i.e., production, is also essentially the same.
The MDR only allows devices with their own UDI-DI to be grouped with a Basic UDI-DI if all of these conditions are met.
You can find a detailed FlowChart in the MedTech Europe guidance for assigning Basic UDI-DI on page 5. This will help you to decide whether you need to assign a new Basic UDI-DI or not. Please also refer to the explanations of the respective decisions on pages 6 to 8.
Interestingly, manufacturers must define the “Medical Device Nomenclature” at the device level (UDI-DI) and not at the product group level (Basic UDI-DI).
This nomenclature appears to be more granular than the “Generic Device Group” and “Category of Devices” introduced by the MDR.
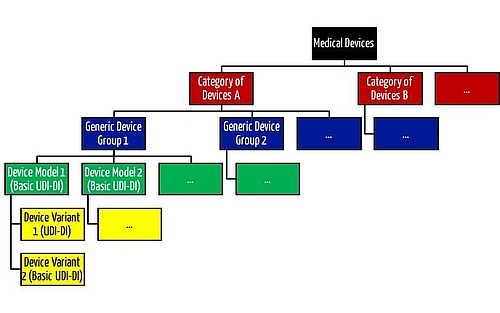
Additional information
Read more about product groups and product categories here.
6. Standards and norms in the context of UDI
In the meantime, there is another nomenclature for medical devices which is intended to be used for EUDAMED. In addition, some standards have been published in the broader context of UDI:
- ANSI/HIBC 2.4:2013 The Health Industry Supplier Labeling Standard: For Patient Safety & Unique Device Identification
- ISO/IEC 15415:2011-2-D Print quality standard
- ISO/IEC 15418:2016 Symbol data format semantics (GS1 application identifiers and ASC MH10 data identifiers and maintenance)
- ISO/IEC 15424:2008 Data carrier identifiers (including symbology identifiers) [IDs for distinguishing different barcode types]
- IISO/IEC 15434:2019 Syntax for high-capacity ADC media (format of data transferred from scanner to software, etc.)
- ISO/IEC 15459 Unique identifiers (note there are six parts to this standard)
- ISO/IEC 16022:2006 Data Matrix bar code symbology specification
- ISO 15223-1 Medical devices – Symbols to be used with medical device labels, labeling and information to be supplied – Part 1: General requirements (more information in the technical article)
- GS1 Guide, which is also referenced by the FDA
- GS1 Guide on MDR and IVDR. If the guidance is not available there, you can download it here.
Some allocation bodies have now published the formats for the UDI (HRI and AIDC). You can download the specifications here as a zip file.
7. Documents that must contain the UDI
The requirements for using the UDI are spread across lots of MDR articles and annexes. This table may serve as a quick overview:
English | German | Article, reference |
Technical Documentation | Technische Dokumentation | Appendix II |
Declaration of Conformity | Konformitätserklärung | Article 19, Annex IV |
Free Sales Certificate | Freiverkaufszertifikate | Article 60 |
Summary of Safety and Clinical Performance (SSCP) | Kurzbericht über Sicherheit und klinische Leistun | Article 32 |
Patient Information / Implant Card | Patienteninformation, Implatationsausweis | Article 18 |
Periodic Safety Update Report (PSUR) | Regelmäßig aktualisierter Bericht über die Sicherheit | Article 86 |
Reports regarding serious incident, Periodic Summary Report | Meldung von schwerwiegenden Vorkommnissen und Sicherheitskorrektur-Maßnahmen im Feld | Article 87 |
Post-Market Clinical Follow-up (PMCF) Update Report | Bericht über die klinische Nachbeobachtung nach dem Inverkehrbringen | Annex XIV Part B |
Field Safety Corrective Action (FSCA), Field Safety Corrective Notice | Sicherheitsrelevante korrektive Maßnahme im Feld, Sicherheitsinformationen | Article 87 |
Repair, Refurbish, Maintenance Report | Reparatur-, Aufbereitungs- und Wartungsdokumente | |
Logistic Documents (Delivery Note, Invoice, Production Order) | Logistik-Dokumente (Lieferscheine, Rechnungen, Fertigungsaufträge) |
Thanks to Martin Tettke from Berlin Cert for this overview
8. The UDI within your quality management system.
Be sure to include the MDR/IVDR requirements for the UDI in your quality management system (QMS). Implementing the UDI impacts various parts of your QMS, such as production (affixing and verifying UDI carriers), the purchasing process, or your system for traceability.
The Medical Device Coordination Group (MDCG) has published a guidance document that addresses how to integrate UDI into an organization's quality management system (MDCG 2021-19). Benefit from this guidance as a checklist or guide for the (correct) integration of all UDI requirements into your quality management system.
9. News (in chronologically descending order)
April 2022
In our Microconsulting, the inquiries about the so-called "Master UDI-DI" are increasing. According to our latest information, the EU is drafting a legal act to make such a "Master UDI-DI" possible. With this Master UDI-DI, highly individualized devices with apparent clinical similarities should be able to be grouped. The problem with these highly individualized products (e.g., eyeglass lenses) is that the high degree of individualization leads to many UDI-DI entries in EUDAMED. This disproportionate number of entries does not provide any regulatory or safety benefit; therefore, the EU has launched this initiative.
The MDCG has already published two Guidances on the subject:
MDCG 2020-18: MDCG Position Paper on UDI assignment for spectacle lenses & ready readers
February 2020
The article on MDR transition periods also addresses transition periods for UDI and UDI carriers.
April 2019: Another publication from the MDCG
The MDCG proposes that the EUDAMED be amended to allow manufacturers to register “legacy products” in EUDAMED without a Basic UDI-DI and UDI-DI:
[The] MDCG considers it appropriate to adapt the Eudamed design to allow the registration of legacy devices in Eudamed in the absence of a (Basic) UDI-DI.
Source: MDCG 2019-05
Another MDCG publication (MDCG 2019-04) addresses when EUDAMED is mandatory. This clarification is necessary because Article 123 is misleading. For more, see the article on EUDAMED.
October 2018: Publications of the MDCG
The Medical Device Coordination Group MDCG published no less than five documents in October, which we would like to briefly present to you here:
MDCG 2018-3 “Guidance on UDI for systems and procedure packs”
The first of the new documents addresses systems and procedure packs. It repeats the definitions of the terms and mentions one exception: If a natural or legal person makes several products (e.g. medical devices) available “together” specifically for a particular customer, this would not be designated as placing on the market.
Other “clarifications” include:
- A natural or legal person who places systems or procedure packs on the market must be registered.
- This person must assign a Basic UDI-DI to this system or procedure pack and store the associated attributes in EUDAMED.
- The systems and procedure packs require their own UDI-DI and UDI-PI.
The MDCG 2018-4 document goes into more detail about the attributes just mentioned.
MDCG 2018-5 “UDI Assignment to Medical Device Software”
Even after reading this publication several times, it is not clear what the point of it is. In the document, it seems as if the authors are letting readers participate in their own learning process. The results are banal. The requirements regarding the UDI for stand-alone software are already formulated sufficiently clearly in the MDR (and IVDR):
- The concept of Basic UDI-DI also applies to software. The Basic UDI-DI groups software with very similar intended purposes.
- A change to the Basic UDI-DI also results in a new UDI-DI.
- A change of the software name also leads to a new UDI-DI.
- The same applies to significant changes to the software such as intended purpose, contraindications, new languages (user languages, not programming languages).
- The UDI-DI may only remain unchanged for minor patches, and only the UDI-PI is to be changed.
MDCG 2018-6 “Clarifications of UDI related responsibilities in relation to Article 16 of the MDR and IVDR”
Article 16 of the MDR or IVDR is entitled “Cases in which the manufacturer’s obligations also apply to importers, distributors or other persons.” In these cases, economic operators (e.g. distributors, importers) are required to comply with the requirements of the MDR that otherwise apply to manufacturers. This includes the requirements for the UDI and registration of these economic operators.
The MDCG states that economic operators are exempt from this obligation if there is a corresponding agreement with the manufacturer and the manufacturer is indicated as such on the label.
It is not entirely clear why this “clarification” is needed. Everything is stated in Article 16(1)a).
MDCG 2018-7 “Provisional considerations regarding language issues associated with the UDI database”
The MDCG notes that essentially only three (of the public) data elements in EUDAMED allow free text. Since one of the goals of EUDAMED is to provide transparency to European citizens, the question arises as to the language in which these free texts are to be stored.
The MDCG proposes that this data be provided in both English and the languages of the countries in which the product is sold. The same applies to the warnings. The MDCG is considering that universally understood symbols could also be considered.
Conclusion
How much these documents really provide “guidance” can be questioned. The documents do not explain what ambiguities actually exist that need to be resolved. Outsiders cannot understand the choice of topics either.
Are these really the biggest problems we have in interpreting the MDR and IVDR? Would it not be more urgent to define the API and the format for the “bulk uploads,” or at least make recommendations for them? How else are manufacturers supposed to convert their IT systems in time to meet the regulatory requirements regarding EUDAMED and UDI?
March 2018 Publications of the MDCG
The MDCG has given some thought to the Basic UDI-DI. The relevant documents are presented above.
- MDCG 2018-1: Guide to Basic UDI-DI and UDI-DI
- MDCG 2018-2: Requirements for a future nomenclature (as for GMDN or UMDNS): It certainly makes sense for this nomenclature to be free of charge. But if the EU is no further along than this publication suggests, it is likely to take some time.
- UDIWG-1: Description of UDI data and data formats in EUDAMED: The data structure is made clearer e.g. you now know whether something should be stored as a string or a number. However, a concrete database schema is still a long way off. For this, the UDI Working Group specifies when to use a radio button and when to use a drop-down list.
- UDIWG-2: Description of Basic UDI-DI and UDI-DI data: Here the MDCG has copied two slides from the UDI Working Group in poor quality. The document is considered official. We have prepared the content for you in a readable and understandable way.
10. UDI from the FDA’s point of view
The FDA requires a unique number for medical devices, the Unique Device Identification (UDI). This may work well for physical products. It is not a problem to affix stickers on the packaging or on the device. But with software? Where would you put a UDI on a virtual product? You could clearly label the DVDs, but that would ban all downloads.

Fortunately, the FDA has clarified the “UDI requirements” to the effect that “unlike most devices, software will only have to exhibit a means of displaying the UDI [...] it can also be within the software itself”.
It is therefore sufficient to display the UDI within the software. However, when it comes to associating this number with a customer (for example, to be able to contact them in the event of a product recall), this alone is not sufficient. A download without authentication / registration is therefore not useful.
Possible solutions: You could assign a unique UDI during registration and enter it in the software to display it under “Help > About,” for example.
11. FAQ about the UDI
11.1. General
- Q: Do the MDR’s UDI transition periods also apply to the allocation of UDIs?
A: No, the UDI transition periods under Article 123 MDR explicitly apply to the affixing of UDIs on the product and packaging levels. - Q: Are the UDIs also required for the MDD conformity assessment procedure?
A: No, the MDD does not require UDIs.
11.2. Basic UDI-DI
- Q: Where exactly must the manufacturer specify the Basic UDI-DI?
A: Registration in EUDAMED, declaration of conformity, brief report on safety and clinical performance, technical documentation, over-the-counter certificates. The Basic UDI-DI is not added on either the product or packaging levels. Thus, no machine-readable format is required for the Basic UDI-DI. - Q: We distribute Class IIb medical devices as non-sterile or sterile- packed. Except for the sterilization, all manufacturing specifications and process steps are identical. Can I assign only one BASIC UDI-DI for these products or are two required – for implants delivered as non-sterile and sterile respectively?
A: In this case, it would make sense to assign a common Basic UDI-DI for the two variants sterile/non-sterile. You specify the information for “labeled as sterile” at the level of the product UDI-DIs in EUDAMED and not at the level of the Basic UDI-DI. - Q: Do all products that are combined into a Basic UDI-DI have to be described in common technical documentation?
A: The MDR does not specify that only one single technical documentation can exist for a Basic UDI-DI. Thus, it should be possible to maintain multiple technical documentations that reference the same Basic UDI-DI.
11.3. Allocation
- Q: If I assign the UDI-DI using GS1, can I define the packaging unit with the first number (e.g. 0 for 1 unit, 2 for 10 units...). Do I have to do this or can I also assign new UDI DIs since I also have a different REF number?
A: This is optional. You can also assign completely new UDI DIs. - Q: What is the process for the accessories of a medical device?
A: Accessories as defined by the MDR require their own UDI. - Q: Our medical device contains wear parts that are replaced regularly. How are the UDIs for such spare parts allocated?
A: Wear or spare parts do not require their own UDI as long as they are not accessories in the sense of the MDR.
11.4. UDI attachment and UDI carrier
- Q: Is it sufficient to apply the data matrix to the products, or is it mandatory to also apply the numerical codes for direct readability? If so, where can I find this specification as a reference in the MDR?
A: The MDR requires that both machine-readable and human-readable forms be specified in Annex VI 4.1: “The UDI carrier (AIDC and HRI representation of the UDI) is affixed to the label or the product itself and all higher levels of the product packaging.” Shipping containers are not considered a higher level of packaging.
If there is a marked lack of space, only the machine-readable form (e.g. Datamatrix) is to be indicated: “4.7. If there are significant problems accommodating both formats – AIDC and HRI – on the label, only the AIDC format shall be used on the label.” - Q: Can both linear barcodes and 2D barcodes be used? Or do you have to choose one type and then follow through consistently?
A: Different UDI carriers and allocation points can also be used for different products.
11.5. The special case of standalone software
- Q: Where must the UDI appear in standalone software, and in what form?
A: According to MDR Annex VI, 6.5.4, the UDI may be specified within the software, that is “displayed in an easily accessible window for the user in an easily readable plain text format, such as in the system information info window or the startup window.” These include, for example, a help menu, an About box or a splash screen. The UDI must be specified in human-readable format only. - Q: What must the UDI PI contain for software?
A: According to Annex VI 6.5.1 of the MDR, software identification is considered a manufacturing control mechanism and is specified in the UDI-PI. Thus, the UDI-PI typically contains at least the software version number.
11.6. The special case of implants
- Q: It is my understanding that the requirement to label the device directly with the UDI carrier only exists for reusable devices that require cleaning, disinfection, sterilization or reprocessing between patient uses. How should non-sterile implants (plates, screws, nails, etc.) placed on the market be viewed in this context? These implants are not intended for reuse, however, reprocessing is of course required prior to first use. Furthermore, these implants are usually sterilized in storage systems, sometimes together with the necessary instrumentation, and made available for surgery. On these systems there are usually a large number of variants of the implants, only some of which are used. The remainder is then sent for reprocessing to be available again for the next operation, just like the instruments for which there is no doubt that direct marking is mandatory.
The non-sterile implants are therefore not reusable products, but they are always reprocessed. I would now say: You would not have to mark them directly with the UDI, since the main criterion is reuse. But this is presumably in contrast to the intention of providing a system for tracking reprocessing cycles?
Now to the specific question: Are non-sterile marketed implants subject to UDI direct marking requirements as described above?
A: In fact, direct labeling is only required for reusable devices that require reprocessing between uses. For implants, the UDI must only be indicated on the direct packaging. However, the MDR requires that the UDI of implantable devices be identifiable prior to implantation (Annex VI, 6.1.3 MDR). This would no longer be the case in this example, so in this constellation presumably only direct labeling would appear to make sense.
11.7 The special case of systems and procedure packs
- Q: If I have a product combination in my range, which is a system under MDR, I have to mark this system with a UDI. Which UDI-PI (from which product) must then be in the UDI on the system’s packaging? The one from the product with the shortest expiration date?
A: The system has its own UDI. Thus, a stand-alone UDI-PI is assigned. This could contain, for example, the “date of manufacture” of the system or a batch number. - Q: What is considered a “convenience kit” for the purposes of UDI (US FDA)?
A: There is a detailed description with several examples in the following FDA guidance document: https://www.fda.gov/media/95120/download
11.8. USA
- Q: No UDI-PI is entered in EUDAMED, what about GUDID?
A: No UDI PIs are entered in the GUDID either. - Q: Is the “American UDI” (GTIN) also applicable for the European Economic Area after the MDR comes into force?
A: There is no American UDI in that sense. GS1 with GTIN is also accredited in the EU under the MDR as a UDI allocation body. Thus, a GTIN for UDI-DI implementation is also possible within the EU.
11.9. Don’t see an answer to your question?
The EU has established a helpdesk.
Johner Institute helps manufacturers meet UDI regulatory requirements quickly and securely. It responds to brief questions in particular in Micro-Consulting regularly and even free of charge.
Change history
- 2022-05-20: Completely revised
- 2021-07-05: Link to EU Helpdesk added

Author:
Luca Salvatore

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.