Generic device group versus device category
The MDR and IVDR use the terms generic device groups and device category without fully defining them.
For manufacturers and notified bodies, it is important to understand what a generic device group is and what device categories are. It's the only way both can prepare for audits and the evaluation of technical documentation.
But take care: The division of devices into device groups and device categories not only affects the “sampling” of these devices.
1. Definitions: What are device groups and device categories
The EU Medical Device Regulation MDR does define the term generic device group. But it does not define what a device category is. The “Guidance Document“ MDCG 2019-13 gives us more clarity, thankfully.
a) Generic Device Group
MDR Definition
The MDR defines a “generic device group” as follows:
Definition: generic device group
“A “generic device group” describes a group of devices with the same or similar intended uses or with technological similarities that can be classified even without considering specific features;”
Source: MDR Article 2(7)
This definition would certainly lead to consistent discussion about the correct classification. Therefore, it is welcome that the Medical Device Coordination Group MDCG wants the generic device group to be understood…
„as the 4th level of the European Nomenclature on Medical Devices (EMDN) (i.e. combination of one letter plus 6 digits) [in respect of the MDR], and in respect of the IVDR as the 3rd level of the EMDN (i.e. combination of one letter plus 4 digits respectively) in combination with the most appropriate IVP code.”
MDCG 2019-13
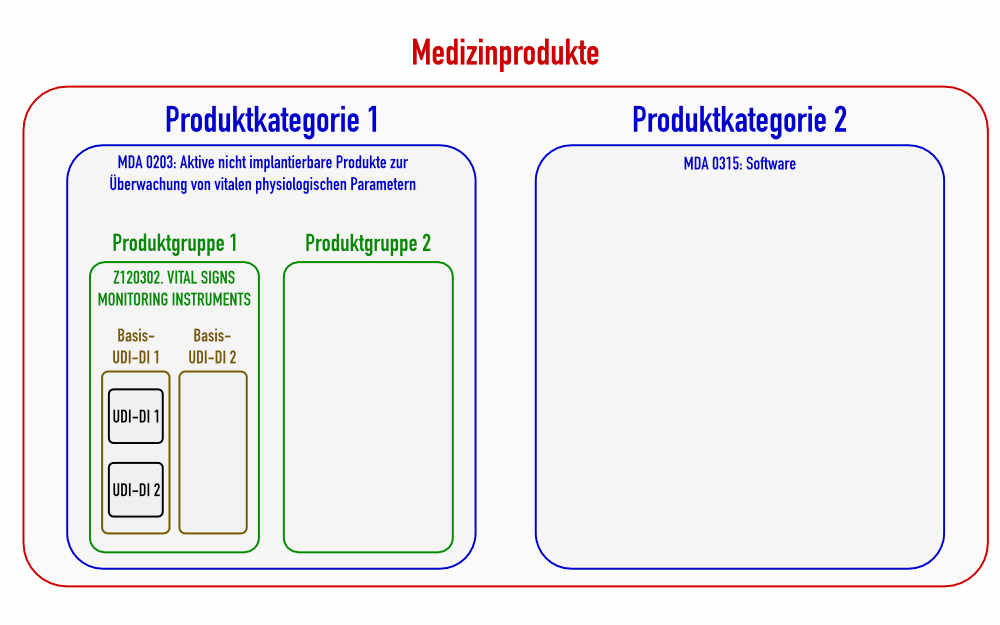
For example, blood pressure monitoring devices are not a generic device group (see fig. 2), because there are already to be put on the 5th level. They are part of the generic device group “Vital Signs Monitoring Instruments”.
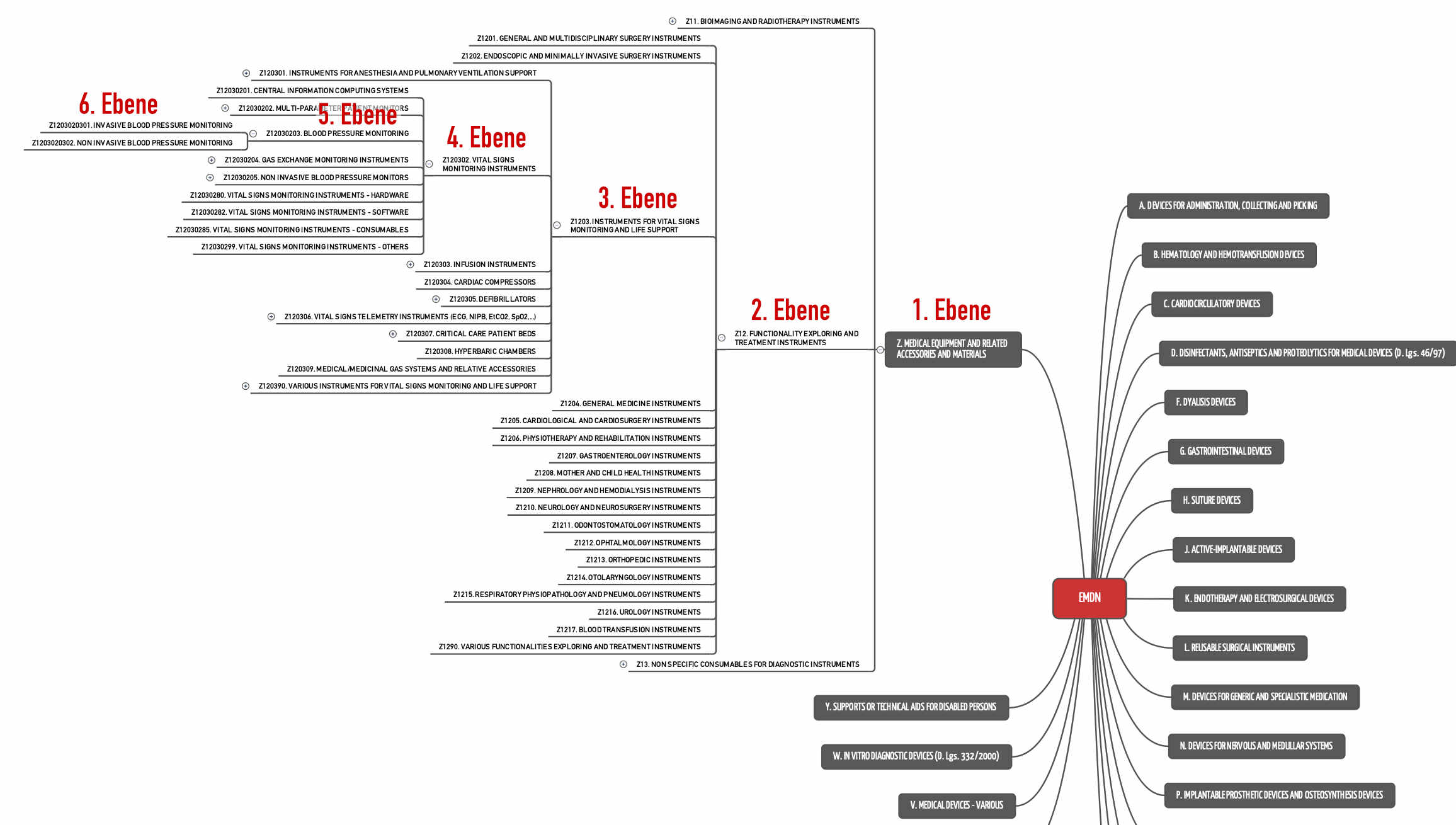
The CND/EMDN system includes 1681 codes on the fourth level - ergo 1681 generic device groups.
Delineation of medical device group pursuant to ISO 13485
ISO 13485:2016 lists the term for “medical device group”:
Definition: Medical device group
“Group of medical devices manufactured by or for the same organization, and which have the same basic design and performance features regarding safety, proper use and mode of action”
Source: DIN EN IS= 13485:2016-08 (3.12)
The term “medical device group” is narrower than the general device groups pursuant to MDR, because it only summarizes devices with the same intended use.
ISO 13485 makes it possible for manufacturers to create common technical documentation for a medical device group. The MDR and MDCG does not see the generic device group as the feature, but the Base UDI-DI.
b) Category of device
The MDR does not have the definition of device category. According to the MDCG, the “codes and the corresponding device types” according to the EU Implementation Directive (EU) 2017/2185 are meant:
Definition: Category of devices
“category of devices should be understood as the relevant MDA/MDN codes (MDR) or IVR codes (IVDR) according to Regulation (EU) 2017/2185 on the codes for the designation of notified bodies.”
Source: EU 2017/2185
This directive differentiates between 71 codes for the devices regulated by the MDR and 79 codes for IVD devices. These categories divide the medical devices into larger classes than the device groups (see also fig. 1).
c) Device Range
The MDCG introduces another term with “Device Range”:
Definition: Device range
“Device range is to be understood as all “device categories“ for Class IIa and Class B devices and all “generic device groups” for Class IIb and Class C devices covered in a certificate.“
Source: MCDG 2019-13
This means that Device Range is an umbrella term, which defines a device category (class IIa) and the generic device group (class IIb) depending on the class of the device.
Additional information
Read more here on the subject of codes, in particular the UMDNS, GMND, CND / EMDN Codes. With Download!
2. Relevance: Why device groups and device categories are important
a) Sampling of medical devices for audits and evaluation of technical documentation
Sampling depends on class
The division of devices into device groups and device categories is relevant to the sampling, meaning the selection of samples for QM audits and the assessment of the technical documentation:
- For less critical devices of class IIa it is enough to select a representative for each device category (article 54/6)).
- For the more critical devices class IIb and III, there must be a representative selected for each generic device group (article 54(4).
Exceptions and limitations of these rules
However, according to MDCG 2019-13, there are limitations and exceptions to these rules:
- If a manufacturer has a large number of devices, at least 15% of these devices must be assessed for each device group or device category within the validity period of the certificate (typically 5 years).
- If a manufacturer only has a few devices, the notified body should still test every device only once during the validity of the certificate. If a device is faulty, the focus should be found, such as through Post-Market Surveillance.
- In the selection of random samples, the notified bodies should not (only) use the random principle, but place priority on devices with new technologies, for example.
- The rule for sampling does not apply to implantable devices of class IIb or devices of class III. For this, the notified bodies must perform the review of the technical documentation for every device.
- Also for "active devices of class IIb that are intended to administer a medicine [...] to the body and/or remove it from the body” there are limitations (article 54(2)).
This also applies to unannounced audits
Sampling also applies to the planned audits as well as to unplanned ones:
Within the framework of unannounced on-site audits, the notified body will test an appropriate sample of the device produced or an appropriate sample from the production process, to determine whether the device produced is consistent with the technical documentation, with the exception of the devices listed in article 52 (8) (2).
MDR Annex IX 3.4
b) Additional scopes of use
How the devices are divided into groups and categories not only has an effect on audits and reviews of technical documentation:
- An EU representative must be designated f or all devices in a generic device group. It is also not possible to designate two EU representatives for two devices in one device group.
- The EU Commission reserves the right to set additional classification rules for device groups and device categories (article 51(3)).
- Depending on device class, the manufacturers must create a “Periodic Safety Update Report (PSUR) “only by device group (article 86(1)).
- Manufacturers must report all of their devices or device groups to the notified body when they make an application for the review of a QM system (annex IX 2.1).
- The certification of the notified bodies must contain the name of the device or device group.
3. Criticism and Conclusion
You may think, finally there is some clarity. It is finally clear how devices are very concretely divided into device categories and then into generic device classes. But it's just not that easy:
The rules for classification of medical devices into categories and groups have apparently been created by various working groups. The result is, that no consistent taxonomy has been created: The codes of the EU Commission for the device categories are not(!) stringently consistent with the higher levels of the CND/EMDN classification.
As soon as manufacturers or notified bodies have to decide to which device category a generic device group belongs, discussion is pre-programmed. It is now time for our authorities to consider a standardization of the standardization.
But let's look at the good thing: We now have two classification systems for the sampling of devices, that compensate for the deficit of lacking or inexact definitions of the terms device category and generic device group.

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.