Market Access of Medical Devices Saudi Arabia
As part of its “Vision 2030,” Saudi Arabia is planning to expand its healthcare infrastructure. Does this offer medical device manufacturers interesting growth opportunities? And will the time and cost of the authorization be worth it?
1. Saudi Arabia, an interesting market for medical device manufacturers?
1.1. A big market
Saudi Arabia has a population of over 34 million. That's about twice the population of Austria and Switzerland combined. However, healthcare expenditure in Saudi Arabia was only 3.3% of GDP in 2006. In Austria, this figure was approximately three times higher (10.3%).
Saudi Arabia's high level of dependence on oil also represents a risk for long-term investments. However, the “Vision 2030” project aims to reduce this dependency.
1.2. Strategic investments in the healthcare market
Part of this initiative explicitly aims to transform the healthcare system. It also aims to promote health tourism. In this context, the Saudi Arabian “medical attaché” in the United States has said:
The government [of Saudi Arabia] is investing a lot of money in healthcare; the budget for just the Ministry of Health is about $10 billion USD. That does not include the National Guard hospitals, Ministry of Defense hospitals and we have 26 universities, most of those have hospitals.
1.3. Conclusion
According to the Global Gender Gap Report, Saudi Arabia is one of worst countries globally in terms of human rights, freedom of opinion or equality. At the same time, Saudi Arabia is a country that already spends a lot of money on healthcare and will continue to increase this spending, making it an interesting market for medical device manufacturers.
However, manufacturers who want to sell their devices in the country first have to meet the regulatory requirements, i.e., they have to have their devices authorized in Saudi Arabia.
2. Registration of medical devices in Saudi Arabia
2.1. Saudi Food and Drug Administration
Medical devices can only be placed on the market in Saudi Arabia if they have been registered with the SFDA (Saudi Food and Drug Administration) and they comply with the Medical Device Interim Regulation. The authority is responsible for the registration and monitoring of medical devices.
2.2. Simplifications and challenges
The SFDA recognizes certain registrations in other countries, which simplifies the authorization process in Saudi Arabia. More on that later.
The SFDA also supports manufacturers by publishing their requirements for medical devices and IVDs on their website and translating (almost) all of their regulations and laws into English.
However, their complexity and the high number of publications regularly cause problems for manufacturers. In recent months in particular, the SFDA has revised numerous requirements in a way that is strongly influenced by the MDR and IVDR.
2.3. The most important laws and guidelines
The Saudi Arabian authority published the most important requirements as the “Medical Device Interim Regulation” in 2009. This regulation contains general requirements for medical devices, and their authorization and market surveillance.
The requirements contained in the “Interim Regulation” are stated clearly in eight “implementing rules”. In addition, the SFDA regularly publishes additional guidelines on specific topics, such as quality management systems, UDIs and software.
You can find an overview of the requirements you need to be aware of in the table below:
Document | Contents |
Medical Device Interim Regulation | General requirements for medical devices and their authorization and market surveillance. N.B.: Also applies to contact lenses and lasers for cosmetic purposes. |
MDS-G5 Guidance on Requirements for Medical Device Listing and Marketing Authorization |
|
MDS-G4 Guidance for Overseas Manufacturers | Summary of requirements for foreign manufacturers |
MDS-IR7 Implementing Rule on Post-Marketing Surveillance | Requirements for the post-market surveillance of devices on the market and reports to the SFDA |
MDS-G42 Guidance on Medical Devices Classification | Classification rules with additional explanations and examples |
MDS-G39 Guidance on Requirements for Reporting of Incidents and Adverse Events of Medical Devices | Reporting deadlines and examples of reportable events |
MDS-IR6 Implementing Rule on Marketing Authorization | Requirements for documentation to be provided or maintained by the manufacturer |
Table 1: Important SFDA requirements
Further Information
The documents named in Table 1 can be downloaded here.
The SFDA is happy to base its registration on existing registrations. Nor does it reinvent the wheel in terms of the documentation and classification requirements – the requirements of the MDS-G5, which the authority published in December 2019, have a lot of parallels with the European MDR and IVDR.
2.4. Slightly different definition of the term medical device
However, the definition of medical devices differs from the definition of the term in Europe, so you should check whether your device falls under this definition – especially if your device only became a medical device under the MDR.
Medical Device: Means any instrument, apparatus, implement, machine, appliance, implant, in vitro reagent or calibrator, software, material or other similar or related article:
Intended by the manufacturer to be used, alone or in combination, for human being for one or more of the specific purposes of:
- Diagnosis, prevention, monitoring, treatment or alleviation of disease,
- Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
- Investigation, replacement, modification, or support of the anatomy or of a physiological process,
- Supporting or sustaining life,
- Control of conception,
- Disinfection of medical devices
- Providing information for medical or diagnostic purposes by means of in vitro examination of specimens derived from the human body;
and which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its function by such means;
Saudi Arabia has not published any of its own guidelines for in vitro diagnostic medical devices. However, these IVD devices are included in the above definition.
Please note!
Products that do not have an intended medical purpose and that are listed in Annex XVI of the European Medical Device Directive must also be treated in Saudi Arabia as medical devices according to the applicable classification rules (MDS-G42, section 4.1).
3. Classification
Devices are assigned to one of classes A to D based on their risk. The class determines the authorization procedure. The next section looks at these procedures.
The classification rules can be found in MDS-G5 and are identical to the 22 rules found in the MDR. You can transfer your classification across accordingly.
Classification in Saudi Arabia | Risk level | Classification according to MDR |
A | Low | I |
A – Sterile | Low-medium | Is |
A – Measuring function | Low-medium | Im |
A – Reusable surgical instruments | Low-medium | Ir |
B | Low-medium | IIa |
C | Medium-high | IIb |
D | High | III |
Table 2: Comparison of classes in Saudi Arabia and according to the MDR
The SFDA has published a guideline (MDS-G42) for the interpretation of the classification rules. The document contains helpful explanations and supplementary examples of how these rules should be applied to devices.
The SFDA also uses European legislation as its base when it comes to the classification of in vitro diagnostic medical device – the IVDR’s seven classification rules can also be found in Saudi Arabia:
Classification in Saudi Arabia | Risk level | Classification according to IVDR |
A | Low individual risk and low public health risk | A |
B | Moderate individual risk and/or low public health risk | B |
C | High individual risk and/or moderate public health risk | C |
D | High individual risk and high public health risk | D |
Table 3: Comparison of classes in Saudi Arabia and according to the IVDR
4. Conditions for authorization
4.1. Condition: authorized representative
Manufacturers without a registered office in Saudi Arabia require an authorized representative (AR) in the country. The AR must register with the SFDA and can then submit medical devices for authorization. The importer, distributor or an independent body can act as the AR.
The tasks of the AR are varied. For example, they are responsible for submitting the application documents and communication with the SFDA. They must also ensure that the device complies with the SFDA’s requirements and, for example, is labeled correctly.
4.2. Condition: QM system
Medical device manufacturers must have an ISO 13485:2016-compliant QM system. The corresponding certificate and the last audit report from the notified body must be submitted to the SFDA.
Incidentally, your AR also has to maintain their own quality management system.
4.3 Condition: MDNR number
The SFDA maintains the online Medical Device National Registry (MDNR). This database lists all devices and Saudi Arabia-based companies. Before a device is placed on the market, the AR submits the relevant information and is assigned an MDNR number.
Please Note!
The MDNR number is not an authorization number. However, it is required for authorization.
The MDNR database itself is not public. However, the SFDA publishes all devices on its website after successful authorization.
4.4. Lower requirements if there is an existing authorization in a GHTF founding country
An authorization or certification in Australia, Canada, Japan, the USA or the EU simplifies the authorization process in Saudi Arabia. Proof of an existing authorization significantly reduces the amount of documentation that has to be submitted.
If you have this proof, you only need to comply with a small number of Saudi Arabia-specific requirements, for example regarding labeling and instructions for use.
Please note!
Submissions through the GHTF-Route will only be accepted until December 31st, 2021. Submissions received after this date will not be accepted and have to follow the TFA-Route (Medical Device Technical File).
The official announcement of the SFDA can be found here.
5. Authorization procedure
The choice of procedure depends on the class of the medical device.
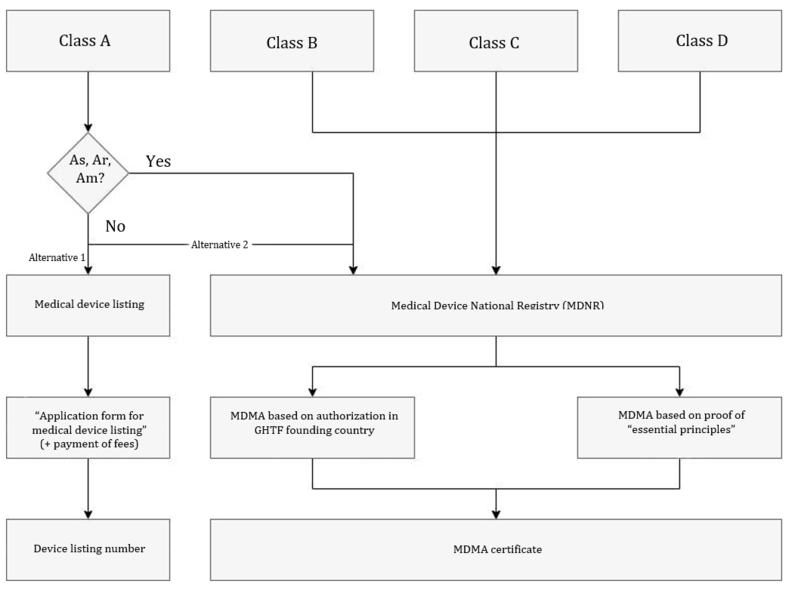
5.1. Listing procedure
Cass A devices can be authorized either via the listing or MDMA procedure. However, the simplified listing procedure is recommended.
Class A devices from the following categories are excluded from this:
- As: sterile
- Am: with measuring function
- Ar: reusable surgical instruments
For the listing procedure, the authorized representative submits the medical device listing application form to the SFDA and pays the fee due.
The SFDA checks the documents submitted and issues a medical device listing number within a few days.
5.2. MDMA procedure
All devices, with the exception of devices listed in section 1 require a Medical Device Marketing Authorization (MDMA). Again, there are two options for the MDMA method:
Option 1: Refer to the authorization or certification in one of the GHTF founding countries.
Option 2: Demonstrate to the SFDA that the “essential principles of safety and performance,” which the authority demands, for example, in the Medical Device Interim Regulation, have been complied with.
Which documents manufacturers need for which procedure is explained below.
The AR submits the documentation to the SFDA. The authority will check your documents and issue the written marketing authorization. This authorization is valid for a maximum of 3 years, depending on the validity of your base certificate. You can find out more about this in the authorization documents section. The SFDA also issues the medical device listing number.
For questions that the AR cannot answer themselves, they can contact the manufacturer.
Please Note!
Manufacturers have to stay on the ball: if the SFDA does not receive a response within 60 days, the submission will be canceled. The fee will not be refunded by the authority. The authorization also has to be renewed at the latest 60 days before it expires.
6. Registration documents
Please note!
As part of the “Vision 2030” it is planned to update the regulatory approach regarding the registration of medical devices. The SFDA intendeds to implement own requirements independent from existing marketing authorizations. SFDA already started to increasingly request the complete technical documentation. Such requests are independent of the classification and can also affect class A manufacturers.
6.1 MDMA based on an existing marketing authorization in a GHTF founding country
The basis for the MDMA procedure is, in this case, the authorization/certification in one of the GHTF founding countries. This procedure has the advantage that manufacturers do not have to submit their complete technical documentation. They can refer to an existing “base authorization.”
The documentation that has be submitted depends on this base authorization: in practice, it is clear that CE marking is taken as the basis for European manufacturers.
Manufacturers must now demonstrate that they meet the requirements of their “base” country. They also have to confirm that they comply with the SFDA's requirements. However, the SFDA does not carry out a full review of the documentation.
Documents to be submitted
- Proof of marketing authorization in the “base country,” e.g., the declaration of conformity (this should be valid for at least another 3 months after the authorization from the SFDA)
- The classification in the home country
- Certificate of free sale
- Proof of the QMS (generally through your certificate and an audit report)
- Information about the device
- Marketing material, labeling and instructions for use
- Information on incidents and / or field safety corrective actions, as well as a commitment to inform SFDA in the event of an incident
- Proof that the product complies with the SFDA regulations:
- Declaration of conformity according to Annex 11 of MDS-G5
- Confirmation that the device fulfills its intended purposes under the specific climate conditions found in Saudi Arabia
- Measures taken to ensure the safe transporting of the device
- The official designation of the AR
6.2. MDMA on proof of compliance with the "essential principles of safety and performance”
In this case, the SFDA will review the technical documentation for at least the following aspects:
- Device description, including variants and accessories
- Specifications
- Marketing material, labeling and instructions for use
- Risk management documents
- Essential principles checklist (this combines chapters I, II and III of Annex I of the MDR)
- Proof of verification and validation
- PMS plan
- Periodic safety update report / PMS report
- Classification according to SFDA rules
- Proof of a QMS according to ISO 13485:2016
- Proof that the product complies with the SFDA regulations:
- Declaration of conformity according to Annex 11 of MDS-G5
- Confirmation that your device can fulfill its intended purposes under the specific conditions in Saudi Arabia
- Measures taken to ensure the safe transporting of the device
- The official designation of the AR
- Confirmation that the SFDA will be informed in the event of incidents or field safety corrective actions
The SFDA reserves the right to request additional documents that prove that the device complies with the Medical Device Interim Regulation.
For the assessment of the documentation, Saudi Arabia has something comparable to the notified bodies in Europe: the conformity assessment bodies (CAB). These CABs are commissioned by the SFDA to review the documents.
All the documents have to be submitted in English.
The labeling can be in English if the device is intended for use by professional users. If it is intended for use by non-medical users, the label must be submitted in English and Arabic.
7. Reporting incidents and recalls
Requirements for the reporting of incidents, corrections and recalls can be found in Implementing Rule 7 and the guidelines MDS-G39 and MDS-G22 that have been published in the last few months.
Reportable incidents, adverse events and field safety corrective actions are defined here, and corresponding examples are given.
The following are subject to mandatory reporting:
- An event outside Saudi Arabia or a field safety corrective action that affects a device registered in Saudi Arabia; or
- An incident in Saudi Arabia.
If in doubt, it is better to report once too many times than once too few.
The SFDA maintains a database, the NCMDR (National Centre for Medical Device Reporting). Your AR will submit the corresponding report for you here.
The following deadlines must be taken into account:
Incident / adverse event | Reporting deadline |
Serious public health threat | 2 calendar days |
Death or severe deterioration of state of health | 10 calendar days |
Other incidents | 30 calendar days |
8. Conclusion and costs
The SFDA has issued extensive legislation and guidelines on medical devices, some of which is based on European legislation. An authorization in a GHTF founding country simplifies authorization.
The authorization procedure, duration and cost of the registration depend on the class of the device.
| Class A* | Class Am, As, Ar | Class B | Class C | Class D |
SFDA fee [SR] | 500 | 30,000–50,000** | 30,000–50,000** | 30,000–50,000** | 30,000–50,000** |
Complexity | 1 | 2 | 2 | 2 | 2 |
SFDA review times | Few days | ~ 1–2 months | ~ 1–2 months | ~ 1–2 months | ~ 1–2 months |
Validity | Max. 3 years |
*except class As, Ar, Am
** depending on the number of devices to be registered
The Johner Institute’s team can help you have your medical devices authorized in Saudi Arabia. We will also be happy to answer your questions free of charge through our micro-consulting service.
-------------------------
Change History
2020-10-08: Adding “Please note” in chapter 6. Adding additional column for class Am, Ar, As devices in chapter 8.

Author:
Margret Seidenfaden

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.