Product Liability: Medical Device Manufacturers Pay Attention!
Neither the MPG nor the MDR contain regulations on product liability, i.e., manufacturers’ liability for harm caused by defective devices.
However, manufacturers should be aware that a lot of legal regulations entitle injured persons to claim compensation if a defective medical device causes harm to a patient, user, or third party. Liability may result, for example, from:
- Tort law (Section 823(1) and (2) BGB)
- Produkthaftungsgesetz (ProdHaftG)
- Damages for breach of duty (e.g., Section 280 BGB together with the contract)
Product liability, which protects consumers (e.g., patients) in the event of product damage, is particularly relevant in practice. This article addresses product liability according to the Produkthaftungsgesetz.
Download
You can download the January 2024 draft of the Product Liability Directive here.
The rest of this article does not yet consider this version of the draft.
1. Product liability: who is liable for what?
a) Who is actually liable in the first place?
The Produkthaftungsgesetz primarily regulates the liability of manufacturers. This includes both the actual manufacturer as well as the legal manufacturer (known as the “quasi-manufacturer”).
The importer is treated in the same way as a manufacturer if the product is imported for the purpose of sale, hire, leasing or any other form of distribution with an economic purpose (Section 4(2) ProdHaftG).
If a manufacturer’s EU authorized representative is responsible for one of these tasks, which is generally the case under the MDR, they may also be liable under the Produkthaftungsgesetz since the ProdHaftG permits more than one manufacturer to be liable at the same time (Section 5 ProdHaftG). This means that, in addition to the actual manufacturer, the EU authorized representative can also be sued for damages, making them jointly and severally liable.
The Produkthaftungsgesetz also regulates supplier liability, i.e., the liability of distributors in cases where it not possible to determine who the manufacturer is. The MDR talks about distributors, the Produkthaftungsgesetz about suppliers.
The labeling requirements of the MDR help suppliers/distributors to identify the actual manufacturer.
The liability claim is usually brought against the legal person, i.e., the company. You can find out when an employee might become personally liable in this article on employee liability.
b) Which damages can be subject to compensation?
The aim of the Produkthaftungsgesetz is to ensure compensation in the case of death, physical injury or health damage or of damage to property, provided that an item of property other than the defective product that was used mainly for private use or consumption was damaged.
Liability for side effects is not clearly regulated. Therefore, in liability claims relating to side effects in the case of medical devices in particular, the product's compliance with the regulatory requirements is checked. In this case, the main focus is on the product’s benefit/risk profile and its instructions for use are reviewed by experts.
2. Produkthaftungsgesetz
a) How does the law define a product?
Only be a movable thing can be a defective product (Section 2 ProdHaftG). It includes all physical and non-physical products, not just medical devices.
The term “product” should not be equated with the term “product” as used in ISO 13485 because this standard uses the term product to refer to services as well as physical objects and software.
a) How does the law define a defect?
The Produkthaftungsgesetz uses the term “defect” in a very broad sense. A defect exists if the product does not offer the safety that you would be entitled to expect taking all circumstances into account (Section 3(1) ProdHaftG).
In practice, proving a defect is key for a judicial decision. Recognized defect types include:
- Design defect: The product design does not meet the required safety standard
- Manufacturing defect: Deviation during production from the standards set by the manufacturer themselves with regard to design and quality
- Instruction defect: Inadequate explanation of how to use the product and the associated risks
The ineffectiveness of a product can also lead to liability, as can justified suspicion of a defect (i.e., potential defect in the case of a defective product series or batch).
Harm attributable to an absence of or deficient product surveillance (e.g., inadequate post-market surveillance (PMS)) is not compensated under the Produkthaftungsgesetz but under general tort law (§ 823 BGB).
A good PMS system helps to demonstrate compliance with these obligations. In addition to the the products manufactured by the company itself, it should include relevant accessories (even if they are produced by a different manufacturer).
Tip
See the "Honda ruling” on this.
3. Additional regulations and judgments
In addition to the Produkthaftungsgesetz, there are a large number of other legal standards and judgments that are referred to for damages claims.
a) Relevant regulations
The following regulations are particularly relevant at a European and national level:
- ProdHaftG
- BGB, particularly Section 823 et seq. BGB
- MDRand IVDR
- MPG/MPDG
- MPSV
- MPBetreibV
Depending on the exact type of product, other laws may apply, for example, the DiGAV if statutory interoperability requirements are not met and harm occurs as a result.
b) Relevant judgments
Case law is also used in judgments. The following judgments are particularly relevant for medical devices:
German Federal Court of Justice (BGH) judgments
- December 09, 1986, ref. VI ZR 65/86 (Honda judgment): Manufacturer's product monitoring obligations, particularly with regard to third-party accessories
- December 16, 2008, ref.: VI ZR 170/07: Obligations of manufacturers in the case of products with safety defects
- February 27, 2020, ref.: VII ZR 151/18 (Tortious liability for consequences of the use of PIP breast implants)
German Higher Regional Court (OLG) judgments
- OLG Saarbrücken, judgment of August 03, 2011, ref.: 1 U 316/10-89: Manufacturer's liability for medical devices - telescopic nails
- OLG Düsseldorf, judgment of March 14, 2012, ref.: I-15 U 122/10: Liability of medical device distributors, in particular warning and recall obligations, MPSV obligations
- Kammergericht Berlin, judgment of April 03, 2014, ref.: 20 U 253/12: Product liability for combinations of medical devices
- OLG Schleswig, judgment of August 29, 2014, ref.: 4 U 21/13): User and operator responsibility for a medical device
- OLG Frankfurt, judgment of January 13, 2015, ref. 8 U 168/13: Prima facie evidence against a medical device manufacturer
ECJ judgments
- March 5, 2015, ref.: C-503/13, C-504/13 (Boston Scientific/AOK Sachsen-Anhalt): Product liability for potentially defective medical device
- February 16, 2017, ref.: C-219/15 (Schmitt/TÜV Rheinland): Obligations and liability of notified bodies with regard to medical devices
- September 6, 2018, ref.: C-346/17 P: Prohibition of the placing on the market of a medical device
Other judgments
- Freiburg State Court, judgment of October 15, 2018, ref.: 1 O 240/10: Claims for compensation resulting from product liability for defective medical device – prosthetic hip
4. Evidence in product liability cases
a) Principle
According to the Produkthaftungsgesetz, the injured person has to prove the product defect, the damage and the causal relationship between the defect and the damage (Section 1(4)(1) ProdHaftG).
The injured person must demonstrate that the product was defective at the time the damage occurred. However, they do not have to prove where and when the defect occurred. Instead, the manufacturer has to prove that it did not breach its duty of care, i.e., it must prove that its product was free of defects at the time it was placed on the market (Section 1(2)(2) ProdHaftG).
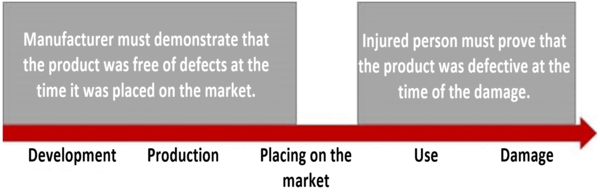
This is often referred to as the reversal of the burden of proof, secondary burden of proof or relaxation of proof. The reason for this relaxation is that the injured person is, as a general rule, not able to inspect the production processes. They, therefore, only have to prove the objectively defective state of the product.
The manufacturer must then subjectively prove that it is not responsible for the fault. It does not have to provide what is known as full proof. (This means that the court does not need to be fully convinced.) It is enough if “under the circumstances it can be assumed” that the product was free of defects at the time it was placed on the market.
b) Examples
Artificial hip
Following a hip replacement, a fracture occurs just one year after implantation.
As the prosthesis is a cast prosthesis, the injured party argues that the fracture was due to a casting error during production (e.g., the formation of a blowhole, which is a predetermined breaking point).
In this case, the manufacturer must prove that there were no defects during the production of the specific prosthesis and that there was no casting error. In practice, this is proven with expert evidence, i.e. by submitting a technical opinion.
Electrocautery unit
During an operation involving an electrocautery unit, the patient and surgeon suffer burns.
The patient argues in the legal action that their burns were due to the inadequate electrical safety of the electrocautery unit.
Therefore, the manufacturer must prove that its product functioned properly and that the state of the science and the art was complied with during production. It can also argue that there was an operator error (use error) and that, therefore, the surgeon is responsible for the injury to the patient.
Expert opinion is again submitted as evidence in this case.
c) Special features of medical devices
The examples show that the product liability has special characteristics in the case of medical devices when compared to other products:
Medical devices are often used by physicians as well as patients, or medical devices only come into contact with patients with the involvement of a physician.
In these cases, in addition to or instead of the product defect(s), there may be a physician error that causes (or contributes to) the damage. So, in the case of a hip replacement, it is also possible that the prosthesis was implanted incorrectly, and the fracture was due to this error by the physician. Differentiating between these alternative causes is a major challenge in practice.
5. Calculating the liability amount
In the case of product defects, the liability amounts vary greatly from case to case. The amounts depend on the damage incurred and must always be calculated individually.
a) Compensation for pain and suffering
In practice, damages are mainly intended as compensation for pain and suffering. The value of compensation for pain and suffering is generally based on:
- The extent of the injuries (intensity of the pain, recovery time, etc.)
- Possible consequential damages
- Impairment of private or professional life
- Age of the injured person
- Level of guilt (degree of culpability, possible contributory negligence)
- Economic situation of the parties involved (income, assets)
If there is permanent damage, higher compensation for pain and suffering must always be expected than in the case of temporary injuries.
Death represents a great loss, but does not generally justify compensation for pain and suffering as this is primarily based on suffering. Only the costs of the attempted cure, property damage suffered, funeral expenses and maintenance costs are reimbursable.
Close relatives (e.g., spouse, partner, parent or child) are also entitled to compensation for the mental suffering inflicted (known as shock damages). In practice, however, this compensation for pain and suffering rarely exceeds €10,000.00.
b) Loss of earnings, household management
The injured person is entitled to compensation for loss of earnings and for household management costs. This applies when a person is unable or only partially able to manage their (family) household due to the injury.
However, the damages are only justified if they are the result of a product defect. In this case, we talk about the contributing cause.
c) Differences with the USA
In the USA, much higher damages are awarded than in Germany. This is due to the differences between the two legal systems.
In additional to the right to compensation for pain and suffering and damages, the US system also provides for punitive damages, which are particularly high. German civil law, on the other hand, is based on a principle of compensation and does not pursue punitive or deterrent objectives. Criminal law aspects only play a role in criminal proceedings.
6. Tips
a) Tips for manufacturers
MDR/IVDR-compliant development is essential to minimize liability risks. This concerns in particular:
- The device's compliance with the general safety and performance requirements
- MDR/ISO-13485-compliant quality management system
- Comprehensive risk management system
- Clinical evaluation
- Systematic post-market surveillance (PMS) for each product
As part of the post-market surveillance, manufacturers must assess all feedback on deviations in terms their severity and probability against the expected risks and document it accordingly in the risk report.
Complete technical documentation that is regularly updated as part of the post-market surveillance (PMS) can make it easier to submit evidence in liability proceedings.
Tip
The scope of the data and information that manufacturers need to collect and evaluate in the context of post-market surveillance is constantly increasing. Therefore, it is advisable to automate these activities as far as possible, as, for example, the post-market radar does.
In addition, manufacturers also have to strictly comply with the notification requirements of the MPSV.
It is advisable to take out insurance (if necessary, with reference to the relevant requirement in the MDR), even if not every risk is insurable. Insurance companies that specialize in medical law can advise the manufacturer on the right tariff.
In the event of a liability case, manufacturers should seek professional legal assistance. They should make sure that the legal advisor is appropriately specialized and has litigation experience.
The process often also depends on the expert, whose opinion the lawyers will examine in detail; or they may have to use another expert for counterevidence. It is often not easy to find an expert to provide an opinion for specific product defects.
b) Tips for distributors
Suppliers or distributors should pay attention to the MDR/IVDR obligations, particularly the obligations relating to labeling, to ensure that they do not become liable as a “quasi-manufacturer” due to incorrect labeling.
The labeling obligations can be found in Article 14 of the MDR paragraph 2 with reference to Article 10 paragraph 11, which in turn refers to the details in Annex I paragraph 23.
If it is not clear from the labeling who the manufacturer of the medical device is, the injured person may request the supplier to name the manufacturer. This request should be complied with within one month, otherwise compensation can be claimed from supplier as the manufacturer under the Produkthaftungsgesetz (Section 4(3) ProdHaftG).
c) Tips for patients
If a patient is harmed due to a medical device defect, it is important they keep good records of the treatment and recovery process and any harm that may have occurred.
The patient should also consult a lawyer who specializes in medical device malpractice. Only an experienced specialist can assess whether a claim has to be submitted against a physician or hospital, or whether they should be notified of the claim.
7. Conclusion, summary
Product liability plays a major role in legal practice and should not be underestimated by medical device manufacturers and other economic operators such as importers and suppliers.
Good documentation of the company's internal processes, particularly the manufacturing and post-market surveillance (PMS) processes, the risk management files and the QM system documentation can play a critical role in liability cases.
The technical documentation of a medical device should also always be complete, correct and kept up to date.
If you need help bringing your procedures and records up-to-date and minimizing the risk of a liability claim, you can contact the Johner Institute (e.g., via the contact form). If you need help with a legal dispute, the specialist lawyer Sonia Seubert will be happy to help you.
Change history
- 2024-02-20: Draft Product Liability Directive of January 2024 inserted at the beginning of the article
- 2021-02-09: Article published

Author:
Sonia Seubert

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.