Market Access of Medical Devices in Japan
Tuesday, May 12, 2020
The authorization of medical devices in Japan represents a big challenge for European manufacturers. But it’s not one they should shy away from, because Japan is one of the 10 biggest markets in the world.
This article will tell you about the requirements you have to meet and the best way to overcome the hurdles in your path.
A) Regulatory framework for authorization
1. Regulatory authorities
The Pharmaceuticals and Medical Devices Agency (PMDA) is Japan's regulatory authority. It is subordinate to the Ministry of Health, Labour and Welfare (MHLW).
The mission of the MHLW is to protect the population of Japan against health hazards caused by pharmaceutical products and medical devices that are not sufficiently safe or effective or not of sufficiently high quality. With regard to the regulation of medical devices, the MHLW's tasks include:
- Registering manufacturers
- Licensing certain actors (MAH, dealers and repair service providers)
- Enacting ministerial ordinances, guidelines and industry standards
- The final release of medical device authorizations
- Monitoring the PMDA
The PMDA, which was created in 2004, is responsible for the actual tasks within the authorization procedures for medicinal products, medical devices and regenerative medicine products for the Japanese market. The PMDA’s most important tasks include:
- Conducting the authorization procedures, including the regulatory review
- Providing advice on clinical studies and authorizations being targeted
- Inspections of manufacturers for compliance with Good Manufacturing Practices (GMP)
- Market surveillance
2. Regulations
Since 2014, the Pharmaceutical and Medical Device Act (PMD Act) has been the most important law. It replaced the Pharmaceutical Affairs Law (PAL).
The PMD Act includes requirements for the marketing, quality assurance and guaranteeing the efficacy and safety of medicinal products, medical devices, regenerative medicine products, cellular therapies, gene therapies and cosmetics.
In the hierarchy of regulations, the PMD Act is followed first by “Cabinet Ordinances”, then “Ministerial Ordinances/Notifications” and lastly “Administrative Notices.” These regulations are relevant to the authorization of medical devices in Japan. “Administrative Notices” are equivalent to MDCG and FDA guidances and are not legally binding.

B) 8 steps to authorization
Step 1: Establish a QM system
As a foreign manufacturer, you do not have to demonstrate “home country approval” to have a medical device authorized in Japan. However, an ISO-13485 certificate will help you a lot when it comes to demonstrating that the Japanese quality management (J-QMS) requirements have been met.
The quality management system requirements can be found in Ministerial Ordinance No. 169 (MO 169), which was revised in 2014. MO 169 is essentially harmonized with ISO 13485:2003 (comparison table, section 2). However, Chapter 3 contains additional requirements that you, as the manufacturer, and your authorized representative (MAH) must comply with. These include requirements for retention periods and vigilance, and rules for communication between the manufacturer and authorized representative.
Step 2: Establish the necessary roles
Marketing Authorization Holder (MAH)
As the next step, as the manufacturer, you have to appoint a local authorized representative, known as the Marketing Authorization Holder (MAH). The MAH assumes liability for your devices in Japan and is the owner of the authorization.
Your MAH not only has to be based in Japan and to submit your marketing authorization documentation, it also has to assume full responsibility for the QM system and, after the authorization of your device, is also responsible for the device or batch release, the post-market surveillance and vigilance.
Not every citizen is allowed to be an MAH. The MAH must first apply for a license (business license known as KYOKA) from the MHLW. The following types of licenses are available:
- First class: the MAH may be appointed for all medical devices (classes I, II, III, IV) (read on for more on classification).
- Second class: the MAH may be appointed for all class I and II medical devices.
- Third class: the MAH may only be appointed for class I medical devices.
An MAH must fill at least the following three rolls:
- General marketing supervisor/director
- Quality manager
- Safety manager
For a third class MAH, one person may perform all three roles. For a second class MAH, the general marketing supervisor can also perform the role of the quality manager or the safety manager. For a first class MAH, the general marketing supervisor must not be the safety manager. Therefore, at least two people are required for first and second class MAHs.
The role of the MAH could, for example, be performed by your distributor, a local office or an independent company. You can read in this article why we don’t recommend having your distributor act as the MAH.
Designated Marketing Authorization Holder (D-MAH)
As an alternative to the MAH, you can name a Designated Marketing Authorization Holder (D-MAH). The difference between the D-MAH and the MAH is that, with a D-MAH, you, as the manufacturer, are the certificate holder. The D-MAH is only responsible for the QMS, including device and batch release, post-market surveillance and vigilance. Therefore, your certificate and device authorization are not affected if you want to change the D-MAH.
Tip
Experience has shown that having a D-MAH is more suitable for small and medium-sized enterprises. In contrast, for larger companies, using their Japanese office or subsidiary as the MAH is the better option. For class I devices, the D-MAH set-up is unfortunately not possible.
Other roles
Lastly, you as the manufacturer also have to name the domestic warehouse manufacturer during the device authorization procedure. This warehouse manager is responsible for storage and shipping in Japan.
You must then specify, before the import, who is responsible for distribution and repair.
These roles (transport, sales, repair) must also be registered with the MHLW.
Step 3: Register as a manufacturer
Before having your medical device authorized in Japan, you must, with the help of the marketing authorization holder (MAH), register as a foreign manufacturer with the Ministry of Health, Labour and Welfare (MHLW). All manufacturing sites responsible for development and final assembly or production must be registered. Component manufacturers do not have to register.
The application for registration is submitted via Form 63-5. The information required for registration includes the following:
- General information (manufacturer's name and address)
- List of all devices
- Declaration by your manager
- Information on the person responsible for regulatory compliance
- Information on the production site
The registration is quite a simple administrative process. It takes about 30 days from submission to registration. The registration is valid for five years and must then be renewed. Changes of, for example, address or contact person must be notified within 30 days.
Step 4: Classify the medical device
The PMD Act differentiates between three types of medical device: general, controlled and specially-controlled medical devices. These are divided into four classes: I (low risk), II, III, IV (high risk).
The following table shows some examples of medical device classifications in Japan.
Class | Description | Example |
I: General medical device | Low risk | Scalpel, tweezers |
II: Controlled medical device | Relatively low risk | Endoscopy, dental alloying agents, ultrasound equipment, MRI units |
III: Specially controlled medical device | High risk | Dialyzer, artificial bone graft |
IV: Specially controlled medical device | Invasive, potentially life-threatening | Stent, pacemaker, artificial heart valve |
Table 1: Classification of medical devices in Japan with examples
In contrast to the EU but like in the USA, devices are classified using predefined product codes: the Japanese Medical Device Nomenclature (JMDN). The JMDN was published in 2005 and is based on the fourth version of the Global Medical Device Nomenclature (GMDN).
The JMDN does not just include the eight-digit JMDN code itself, it also contains:
- The general name of the device type
- Definitions
- The risk-based classification according to the GHTF
- Information on whether the QMS requirements have to be complied with
- The classification rules
- Applicable authorization and review criteria (e.g., applicable standards)
If no suitable JMDN code is available, a risk-based classification based on GHTF rules should be used.
Step 5: Select the authorization procedure
In addition to the classification, there are other factors that influence the choice of authorization procedure.
- Comparator devices in Japan: a distinction is made between generic/me too, improved and novel devices. Generic means that there is already an identical or essentially similar comparator device available on the Japanese market (“substantially equivalent” to a “predicate device”). Improved devices are medical devices for which there are no suitable predicate devices. A novel device is a new type of medical device that is significantly different in terms of its intended purpose, technology, mode of action, or performance.

2. Certification standard: certification standards are specified for a large number of JMDN codes. In this case, authorization can be granted via a third-party review by an independent certification body (see below). For example, for the JMDN code “36208000 – Mobile ultrasound imaging system,” the applicable certification standard would be JIS_T_0601-2-37, which corresponds to IEC 60601-2-37.
Based on the classification and the aforementioned two influencing factors, you can then choose one of the three authorization procedures.
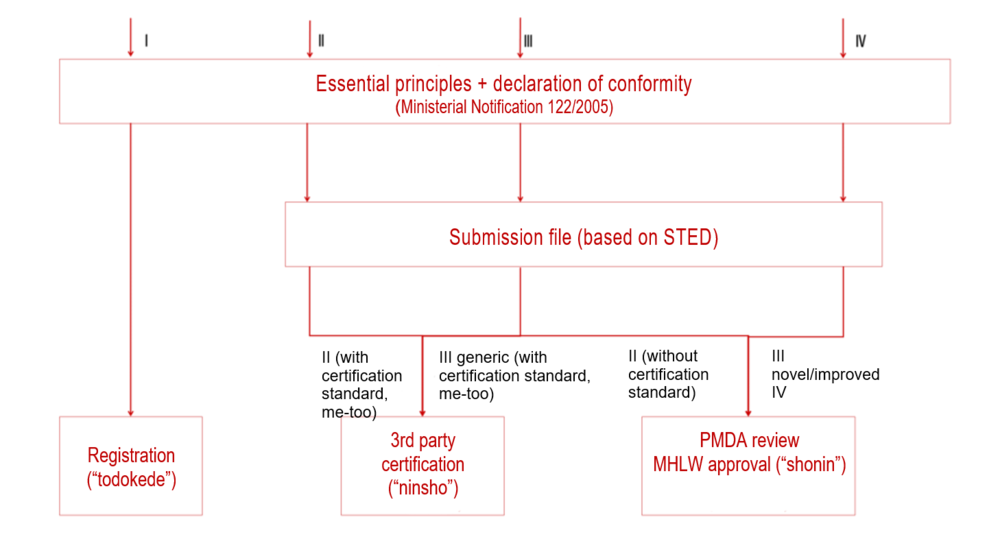
Name of the procedure | Applies to | Short description of the procedure |
TODOKEDE | Class I | Simple registration of devices with declaration of conformity (similar to class I in the EU) |
NINSHO | Class II and III generic, with certification standards | An RCB (Registered Certification Body), as a “third party,” reviews the authorization documents and conformity with the “essential principles.” |
SHONIN | Class II and III without classification standards and class IV | PMDA reviews the authorization documents and conformity with the “essential principles.” |
Table 2: Authorization procedures in Japan
Todokede
The MAH notifies the PMDA of the class I device on behalf of the manufacturer. In this notification, in addition to the manufacturer’s details (manufacturing sites, manufacturing process, registration number), a general description of the medical device (including intended purpose, trade name, mode of action, performance characteristics, specifications, requirements for storage and shelf life), the MAH must submit the instructions for use in Japanese. In addition, you as a manufacturer must always comply with the essential principles. However, there is no verification of compliance in the todokede procedure. The registration is typically completed within a month.
Ninsho
In ninsho procedure, also known as a pre-market certification, the Registered Certification Bodies (RCB) are responsible for reviewing the documentation. RCB are Japanese “notified bodies,” who are responsible for third party review authorization procedures.
The RCB reviews the authorization documents for compliance with the certification standards and the essential principles and, if they do comply, issues the certification. The RCBs include the TÜV Süd, TÜV Rheinland, BSI Group, SGS and DEKRA.
The ninsho procedure applies for most class II devices and some class III devices. On average, the review of the marketing authorization documents by RCBs (for class II and class III devices) takes four months.
Shonin
If you want to have class II and III medical devices without certification standards or class IV devices authorized in Japan, you must go through the strict shonin procedure, also known as a pre-market approval (PMA), with a review and authorization by the PMDA. For most standalone software devices (SaMD), there are currently no certification standards available, so the shonin procedure must be used.
In general, approval standards or approval guidelines have been defined for generic/me too devices. They have been assigned to the individual JMDN codes in the JMDN database. They contain, for example, applicable standards or device-specific guidances.
The shonin procedure usually lasts approximately 12 months, without taking into account the time needed to prepare the submission documents or conduct clinical studies.
The official costs of the Japanese authorization procedures can be found in this list.
NB!
Class I and II devices for which no JMDN code has been defined yet also have to go through this procedure.
Step 6: Compile and submit authorization documents
Prepare the clinical evaluation (may require a clinical study)
As in Europe, before your medical device is authorized in Japan, you must provide evidence of the clinical benefit, performance and safety of your devices in a clinical evaluation.
Japan, like Europe, allows this proof to be provided, under certain circumstances, based on literature data. No clinical evidence has to be submitted for generic/me-too devices. In contrast, clinical studies are required for novel medical devices. For improved devices whose safety and performance have not been sufficiently demonstrated by pre-clinical data and literature data, data from clinical studies also has to be submitted.
It is not always mandatory to conduct these clinical studies in Japan or to use clinical data from Japan.
Tip
It is advisable for your MAH to discuss the recognition of foreign clinical studies with the regulatory authorities in advance.
General authorization documents
All documents submitted during the authorization procedure must be in Japanese.
The submission documents are based on the IMDRF’s internationally recognized STED format. In addition to the STED summary documents, the following annexes are required:
A – Development history (previous device versions, global authorizations)
B – Product specifications
C – Data on stability and shelf life
D – Compliance with the applicable standards and essential principles
E – Performance test data
F – Risk analysis
G – Manufacturing (process, supervision, sterilization)
H – Clinical data
In practice, there is open communication with the authority once they have started reviewing the documents. This means that the scheduled duration of the document review is not interrupted if the authorities have queries.
This interactive communication also enables clinical trials to be authorized relatively quickly.
Marketing strategy tip
As a manufacturer, you would be well advised to plan your marketing strategy for Japan early on. This would mean you were able to present your product at trade fairs even during the authorization procedure.
You can sign a contract with the distributor for this, but do not sell the device until you have received authorization.
Step 7: Request a QMS inspection
Before release of the device, the PMDA or the RCB (depending on the authorization procedure) will audit the MAH's QM system as well as the QM system at the registered manufacturing sites, i.e., the foreign manufacturer's site as well. An independent audit is performed for each device family. The application is submitted by the MAH. The audit takes the form of either an on-site inspection or a document audit. Which procedure the authority decides on depends, among other factors, on whether the site has ISO 13485 certification, on the results of the previous audits, on reported incidents and recalls, and on the complexity of the manufacturing process. For software manufacturers, a document audit is usually carried out.
In a document audit, the regulatory authority checks the following documents:
- Manufacturing site overview
- Organigram
- QM manual
- List of QM documents
- Manufacturing process incl. validation
- MAH contract and vigilance procedure
- If requested: summary of the medical device dossier
Japan has been part of the Medical Device Single Audit Program (MDSAP) since 2015 and has accepted MDSAP audit reports since the end of the pilot phase. Therefore, in general, no additional documents have to be submitted. Manufacturers who undergo an MDSAP audit can thus avoid redundant audits.
Step 8: Plan the post-market phase
Modification of medical devices after authorization
As in many other countries, as a manufacturer you must submit a “change notification” if your devices or other information change. In Japan, the authorities differentiate between partial changes and minor changes. Partial changes include changes to the intended purpose or other changes that affect the safety or efficacy of the device, e.g., changes to the operating principle, materials or user interface. Minor changes covers all changes that do not fall under the definition of partial change. For minor changes, a simple notification within 30 days after the change is made is sufficient. For partial changes, a “partial change application”, i.e., a new submission, is required.
Post-market surveillance
The obligation to perform post-market surveillance also affects your MAH. Japanese regulations make the MAH responsible for reporting incidents involving medical devices to the manufacturer and the regulatory authorities. The requirements are set out in the “Good Vigilance Practice” ordinance (Ministerial Ordinance 135).
Vigilance tip
As a medical device manufacturer, you are obliged to support your MAH in the post-market surveillance. Having corresponding regulations in the contract with your MAH or within the framework of your QM system is strongly recommended.
Section 4 of the MDSAP Companion Document provides a good overview of the requirements for reporting incidents and recalls. It also references the relevant regulatory requirements. In general, you must inform your MAH immediately about serious incidents (including those that happen abroad).
C) Authorization support
The Johner Institute can support medical device manufacturers having their devices authorized in Japan in the following phases:
- Classifying devices
- Selecting the authorization procedure
- Preparing and reviewing authorization documents
- Finding an MAH
Get in touch with us via this contact form if you want your device to be successful on the Japanese market as quickly as possible.
D) Conclusion, summary
The hurdles to the authorization of medical devices in Japan are no longer as insurmountable as they used to be. The MDSAP procedure also helps with this. In addition, there are a lot of parallels with the authorization procedure in Europe and the USA.
However, the roles, such as the MAH (or D-MAH) and the DSM, and the obligation to submit the documents in Japanese require a significant effort.
As a result, the Japanese market is large and stable. According to BVMED, Japan is already eighth highest on the list of countries that German medical device manufacturers export to.

Author:
Luca Salvatore

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.