Supplier evaluation – supplier selection – supplier audits
The MDR and ISO 13485:2016, just like the FDA, set out clear requirements regarding supplier evaluation, supplier selection and supplier monitoring.
This article not only gives you an overview of the regulatory requirements. It also gives you tips on how to implement them and tells you when a supplier audit is necessary.
1. Basic principles of supplier management
a) Examples of suppliers and delivered products and services
As soon as manufacturers stop developing something themselves and start buying it in, they require a supplier evaluation. Examples of products and services supplied externally are:
- Product development
You order the development of an entire product. A special case would be if this product is a medical device. - Component development
You order the development of part of a product. Here too, if this part or component is part of a medical device, it is to be considered a special case. - Component purchasing (“catalogue goods”)
You use a “ready”, that is, an already existing product within your product or medical device as the case may be. - Tool purchase or hire
You buy or hire products as tools. This includes external software as a service, e.g. in the sales department. - IT services
You use an IT service like server hosting and a cloud service. Here, it would be necessary to determine whether this service is a part of your products or services.
b) Supplier selection, supplier evaluation, supplier monitoring
First of all, manufacturers should establish criteria by which they assess the suppliers. Then they carry out the supplier evaluation. Based on this supplier evaluation they select the most suitable supplier/s (supplier selection).
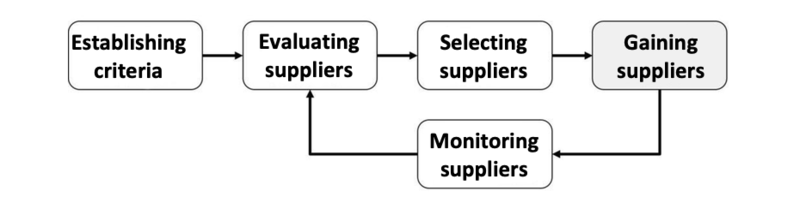
Fig. 1: Supplier evaluation, supplier selection and supplier monitoring is an ongoing process.
Manufacturers monitor suppliers continually, e.g. within the scope of the supplier audit and evaluate the suppliers regularly, for example, based on audit results and the quality of the products and services delivered.
2. Regulatory requirements for supplier management
a) MDR
QM system requirements
The MDR makes it unequivocally clear that quality management must regulate “selection and control of suppliers and sub-contractors” (Article 10 (9)d.). The notified bodies must check that this actually happens.
Supplier audits
The notified body must decide whether a specific supplier or sub-contractor audit is necessary (Annex VII 4.5.2.a, Annex IX 2.3 and 3.3). If this applies, even the suppliers (“sub-contractors”) are subject to unannounced audits – “at least once every five years” (Annex IX 3.4).
The notified body is obliged to take samples of the documentation from the supplier (“sub-contractor”), particularly if the delivered parts have an influence on the conformity of the products and the manufacturer is unable to demonstrate sufficient control over its suppliers (Annex VII 4.5.2).
Product documentation requirements
The manufacturers must specify which suppliers and sub-contractors are involved in development and production (see Annex II, 3.c.).
b) ISO 13485:2016 and ISO 9001:2015
ISO 9001:2015 and ISO 13485:2016 place concrete requirements on the selection and evaluation of external suppliers of products and services – supplier selection, supplier evaluation and supplier assessment. Manufacturers must...
- Establish criteria for the providers/suppliers (examples of criteria are mentioned below)
- Evaluate providers/suppliers according to these criteria
- Select providers/suppliers according to these criteria
- Monitor providers according to these criteria
Heads up!
Please bear in mind that these criteria must be established specifically for the product.
Alongside suppliers, the regulatory requirements also concern products and services respectively. Manufacturers must...
- Establish specifications for the products to be purchased
- Provide the providers/suppliers with the necessary information in writing
- Establish what procedures*, processes and tools are to be used to test the delivered products
- Test the products according to these specifications.
ISO 13485 adds aspects that are specific to medical devices such as:
- regulatory requirements
- analysis of the effects of the product/service purchased on the safety and performance of the medical device
- risks, which are generally assumed for the medical device (regardless of the product purchased)
c) ZLG requirements
You can find further requirements on supplier assessment in the ZLG documents, e.g. documents 3.9 B16 and 3.9 B 17.
d) FDA: 21 CFR part 820
The FDA mentions practically identical requirements in 21 CFR part 820.50 “Purchasing Controls”. Contrary to ISO 13485, it explicitly mentions a quality assurance agreement:
Purchasing documents shall include, where possible, an agreement that the suppliers, contractors, and consultants agree to notify the manufacturer of changes in the product or service so that manufacturers may determine whether the changes may affect the quality of a finished device.
FDA 21 CFR part 820.50
3. Evaluating suppliers
You shouldn’t decide how you select and evaluate your suppliers in every new case, but you should establish a procedure specification for selecting and evaluating suppliers.
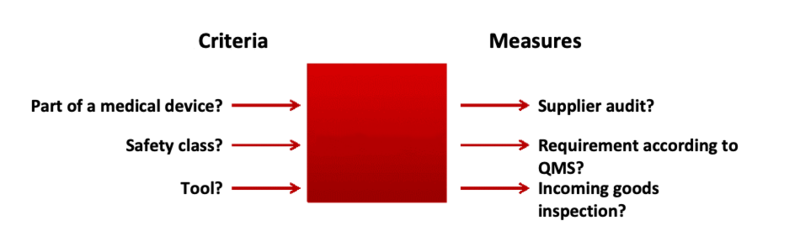
Fig. 2: The supplier control measures, as well as supplier monitoring and supplier evaluation, should depend on specific criteria
In order to fulfill the above-mentioned requirements, this procedure specification must determine criteria and methods for selecting and evaluating suppliers.
a) Step 1: establish criteria
The criteria you can consider when implementing measures for selecting and evaluating your suppliers include:
- Does the supplier develop a medical device or parts/components for it?
- Does the supplier provide services that form part of your services? Such an example is a hosting service provider with whom you offer your software as a service.
- Is your supplier ISO 13485 certified?
- How dependent is the manufacturer on the supplier? Are there alternative suppliers, products or procedures?
- Do you have experience with the supplier in terms of adherence to delivery deadlines and quality of the products delivered?
A Google search that associates the supplier with terms like “problem” or “unreliable” often provides new insight. Product reviews can also be helpful. - Is the product or service business-critical?
Would failure to meet requirements lead to a breach of the law, a breach of data security, disclosure of company secrets, loss of reputation or financial deficits?
If the delivered product is or contains software, further criteria are to be taken into account for the supplier evaluation:
- What safety class does this software belong to?
- Is it SOUP or OTS?
- Does this software itself contain SOUP?
- Is the software a tool or part of a product?
- Is this a case of purchasing, hiring or development?
b) Step 2: list measures for supplier evaluation
Regardless of the criteria, adopt one or more of the following measures:
- Conclude a quality assurance agreement (y/n)
- Adapt the contents of a quality assurance agreement
- Standards to be met by your supplier
- List of procedure specifications that your supplier must follow
- Number and qualifications of staff to be provided by the supplier
- Supplier’s assent to supplier audits including scope and frequency
- Limit potential suppliers to those who are ISO 13485 certified (y/n)
- Inspect incoming goods
- Frequency, sampling
- Methods, e.g. additional tests, visual inspection
- Type and scope of the documentation made available to the supplier, e.g.
- Product specifications
- Acceptance criteria
- Project specifications such as time and budget
- Quality assurance agreement (see above)
c) Step 3: correlate measures and criteria
You certainly won’t be using the methods and criteria mentioned for every supplier. It doesn’t make much sense to subject your stationary supplier to an audit. If, however, your supplier writes the software for your medical device and is not ISO 13485 certified, it is your duty to arrange a supplier audit.
Thus, in the last step you establish which supplier evaluation measures you are to implement and under what criteria. As the rules and regulations can very quickly become confusing, you can group together the measures and stipulate different types of suppliers.
Thus, there could be a category for “highly critical suppliers” with whom you sign a quality assurance agreement and who allow for audits, a full incoming goods inspection and personnel with a certain level of qualifications.
You can set out these rules for supplier evaluation in a table, in a text or as a flow chart.
4. Supplier audits
As explained above, supplier audits are included in the measures that manufacturers take within the scope of ongoing supplier monitoring and evaluation.
Whether and when supplier audits are to take place depends on the criticality of the products and services delivered, as well as whether the suppliers have their own QM system or not.
a) Supplier audit: if the supplier does not have their own QM system
In this case, the manufacturers declare their own quality management system and its rules respectively to be binding for their suppliers.
Manufacturers must check that suppliers are adhering to these rules by means of supplier audits. Within the scope of such an audit, manufacturers check, for example, whether or not the supplier documents development or production according to the manufacturer’s specifications. These audits should be performed at least once a year.
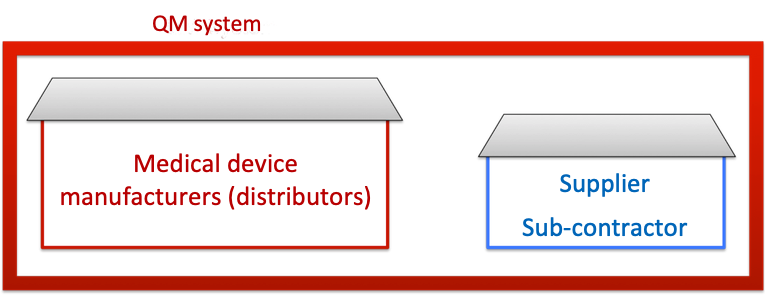
Fig. 3: If the supplier works under the umbrella of the manufacturer’s QM system, during the supplier audit the manufacturer must check their conformity with the QM system.
The manufacturer is also audited. According to ISO 13485 these audits by notified bodies must also extend to suppliers, meaning that it is possible that the auditor may pay the supplier a visit.
As component manufacturers and development service providers do not bring any medical devices into circulation themselves, they do not need to be subjected to any audits by notified bodies. They normally only allow this to meet the requirements of their customers, the manufacturers.
b) Quality management system instead of supplier audits
To prevent their own supplier audit from getting out of hand, many manufacturers prefer suppliers who have their own QM system. In this case, audits on the manufacturer carried out by notified bodies are limited to document inspections.
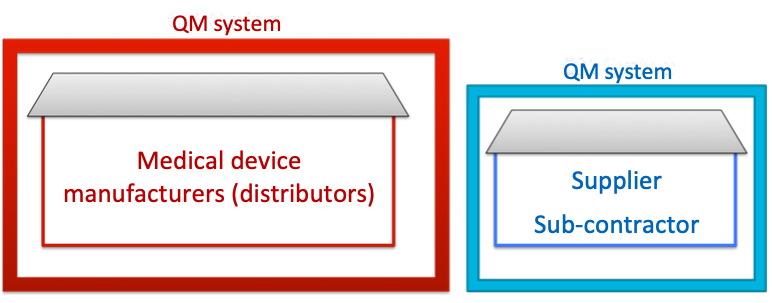
Fig. 4: If the supplier has their own QM system (according to ISO 13485:2016), the manufacturer may refer to that
In the selection of suppliers, above all companies with ISO 13485 certification and not just ISO 9001 lend themselves to medical device manufacturers.
However, even with this type of company, an additional supplier audit is also recommended. Such audits must be performed as a part of the contracts between the medical device manufacturer and the supplier.
c) Which companies can be excluded from supplier audits?
Conformity assessment procedures refer to the development and production of medical devices. This means that whenever a manufacturer has components developed or produced for their medical devices, these work steps may be subject to a supplier audit.
This is different for components that are not specially developed or produced for the medical device such as monitors, mains adapters or off-the-shelf software components. In this case the manufacturers will ensure, within the scope of risk management, that these “purchased parts” (“catalogue goods”) do not lead to any unacceptable risks. A supplier audit would not be carried out there (or be allowed).
Read more on the subject of audits here.
Further information
Read more on the subject of audits here.
a) Supplier evaluation and selection
Manufacturers must evaluate and select suppliers before commissioning them. This choice must be made based on clear criteria.
Supplier control, which particularly includes monitoring the suppliers, is an ongoing process.
The selection of these criteria and the intensity of this control must be risk-based.
b) Supplier audits
Supplier audits are carried out at companies to which part of one’s own tasks, such as development, have been outsourced. Here we often refer to the “extended workbench”. The audit must then be performed according to the rules of the manufacturer’s QM system (ISO 13485).
The manufacturer (distributor) can only spare themselves this audit if the development partner has their own ISO 13485 QM system and presents the corresponding documentation for the product to the manufacturer. The same applies to audits by the notified body.
c) Conclusion
Manufacturers are increasingly outsourcing tasks like development and production, either wholly or in part. The regulations make it clear that by doing so the tasks may not be withdrawn from a quality management system. For this reason, the notified bodies are obliged to also inspect the suppliers, if necessary, and in some cases within the scope of unannounced audits.
So manufacturers are well advised to select and monitor manufacturers with whom they can guarantee consistent quality management and therefore product conformity and safety.
Support from the Johner Institute
The Johner Institute supports manufacturers and supplies in the following tasks, among others:
- Drawing up MDR, ISO 13485 and FDA-compliant procedure specifications for evaluating, selecting and monitoring suppliers
- Formulating quality assurance agreements
- Preparing audits by manufacturers and notified bodies
- Guaranteeing the correct interplay of tasks (e.g. supplier monitoring, risk management, market surveillance, trend analysis, etc.) within the scope of post-market surveillance (central requirement of the MDR)

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.