South Korea: An interesting market for medical device manufacturers
A German medical technology industry analysis from 2020 estimated the market for medical devices in South Korea at USD 6.7 billion. Due to an annual growth rate of imports of (estimated) 10% and the increasing aging of the population with a simultaneous increase in basic medical care, the South Korean market is expected to grow continuously.
This article highlights the regulatory framework that must be adhered to when selling medical devices in South Korea and the hurdles that must be overcome.
Regulatory framework
a) Responsibility
Since 2017, the MFDS (Ministry of Food and Drug) has been responsible for the regulation and approval of medical devices in South Korea. The MFDS is responsible for the registration of medical devices, the definition of regulations, and the issuance of certificates.
b) Regulations
The legal framework for the registration and distribution of foreign medical devices and in vitro diagnostic devices in South Korea is provided by the Medical Device Act and the Act on In vitro Diagnostic Medical Devices. The legal texts serve the following objective and purpose (taken from the English translation):
- The purpose of this Act [Medical Device Act] is to promote the efficient management of medical devices and further contribute to the improvement of public health by providing for matters concerning the manufacturing, import, distribution, etc. of medical devices. (Medical Device Act).
- The purpose of this Act [Diagnostic Medical Devices] is to improve public health and contribute to the advancement of in vitro diagnostic medical devices through ensuring safety, improving quality, and strengthening international competitiveness, of such devices by providing for matters necessary for handling, such as manufacturing and import, and management of, and support for the devices (Act On In Vitro Diagnostic Medical Devices).
c) Medical device qualification in South Korea
The definition of a medical device can be found in Article 2 of the Medical Device Act. This defines a medical device as follows:
The term "medical device" in this Act means an instrument, machine, apparatus, material, software, or any other similar product specified in the following subparagraphs as one used, alone or in combination, for human beings or animals:
- For the purpose of diagnosing, curing, alleviating, treating, or preventing a disease
- For the purpose of diagnosing, curing, alleviating, or correcting an injury or impairment
- For the purpose of testing, replacing, or transforming a structure or function
- For the control of conception
Note on the differences to the MDR
There are several differences with Regulation (EU) 2017/745 on medical devices (MDR):
- The extension of the term to include use in animals (the MDR only includes humans).
- The main pharmacological or immunological effect of the device (described in the MDR) is not included in the Korean definition of the term.
- Obtaining information through in vitro studies is not part of the Korean definition of "medical device."
For definitions of in vitro diagnostic devices, see the Act on In Vitro Diagnostic Medical Devices.
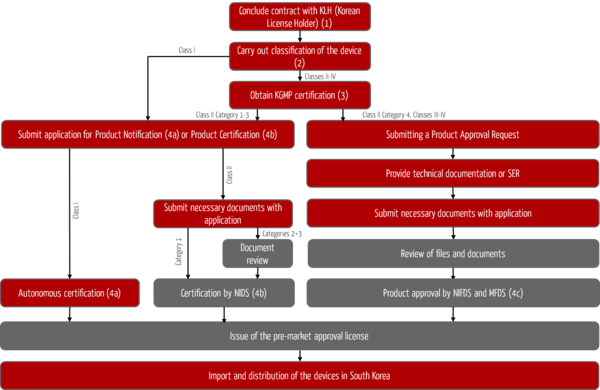
The steps to product registration in South Korea
The registration process for foreign medical devices in South Korea is shown in Fig. 1. The objective of this process is the approval or issuance of a pre-market approval license for your devices by the South Korean authorities and the associated possibility of importing and distributing your devices on the South Korean market.
Attention
Pre-market approval licenses obtained must be renewed every five years.
Step 1: Designation of the Korean License Holder KLH
Before starting the registration process of their medical devices, manufacturers without a branch in South Korea must appoint a Korean License Holder (KLH) in the country and conclude a contract with him. The KLH acts as the legal representative to the Ministry (MFDS).
Korean License Holders must meet the following requirements:
- Be a resident of South Korea and have a registered office in South Korea
- Be registered as a KLH at MFDS with a license for medical devices and in vitro diagnostic devices
- Be registered as a quality manager at MFDS
Notes
The Korean Licence Holder is required regardless of the risk class of the medical devices or in vitro diagnostics.
Together with our local partner, we support you as KLH and with all questions concerning the local medical device regulations. Do not hesitate to contact us.
Step 2: Classification of the medical device
The MFDS uses a four-level classification system for medical devices based on the risk to the human body. It follows the approach of the Global Harmonization Task Force (GHFT) Principles of Medical Devices Classification or the analogous IMDRF rules.
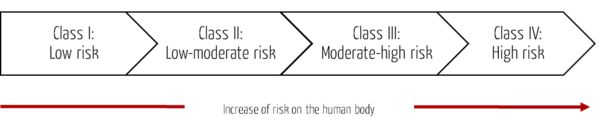
Class | Risk | Example |
Class I | Medical devices with low risk | Forceps, stethoscopes, patient bed (manual operation) |
Class II | Medical devices with low to moderate risk | Syringe, infusion pump, patient bed (electric operation) |
Class III | Medical devices with moderate to high risk | Anesthesia system, ventilator, dialysis system |
Class IV | Medical devices with high risk | Heart valve, coronary stent, bypass systems |
Tab. 1: The classes and risks of medical devices in South Korea
As a first aid for the classification of devices, the database Medical Device Products and their Assigned Classes of the MFDS can be used. At regular intervals, the MFDS publishes an updated overview of classifications of registered medical devices in the database, including examples, descriptions, and category numbers.
Tip
Benefit from the classification rules of the GHTF guideline Principles of Medical Devices Classification (chapter 8.0) as a further aid to classify your devices into one of the four risk classes.
Step 3: Certification according to Korean Good Manufacturing Practices
Foreign manufacturers of medical devices (including in vitro diagnostics) must have their QM system certified with regard to KGMP (Korean Good Manufacturing Practices) before the start of the respective product registration.
KGMP certification is intended to ensure that medical devices are produced, controlled, and followed up in accordance with the requirements and quality standards of MFDS. Detailed information on the implementation of KGMP is provided in the Regulation on Good Manufacturing Practices (GMP) for Medicinal Products.
Class II to IV devices are affected by KGMP certification. Class I medical devices are exempt from this obligation.

The application for KGMP certification is submitted to the MFDS by the Korean License Holder (KLH) chosen and contracted by the foreign manufacturer of the medical device.
Attention
KGMP certification is linked to the selected KLH (Korean License Holder). When changing the KLH, a renewal of the KGMP certificate is necessary.
In the case of new registrations, the inspection is carried out as an on-site audit of the production facility. The costs range from USD 4,000 to USD 6,000, depending on the scope. A lead time of several months is to be expected here.
In the case of product registration with a valid KGMP certificate or when the certificate is renewed, a document audit is carried out. The costs are between USD 1,000 and 1,500. A duration of several weeks is to be expected.
The KGMP certificate remains valid for three years and must then be renewed.
Attention
Despite being based on ISO 13485, certification according to ISO 13585 does not replace the KGMP certificate. Additional requirements must be met or implemented, e.g., post-market surveillance and reporting.
Step 4: Product registration
The "approval procedures" of medical devices in South Korea (also defined as registration or certification of medical devices) differ depending on the defined medical device class (I to IV). The different procedures are described in more detail below.
Attention
All documents and necessary information to be prepared for registration, certification, or approval of your devices must be provided in Korean!
Registration of class I devices (4a)

Manufacturers of class I medical devices submit an application for product notification to the NIDS (National Institute of Medical Device Safety Information) for the registration of their devices.
After successfully uploading the complete application, including all information, the manufacturer can independently complete the product registration of its class I medical devices. There is a cost associated with this process of approximately USD 75.
Note
Class I medical devices are exempt from KGMP certification.
Certification for class II devices (4b)
The approval or certification procedures of class II devices differ in terms of Substantial Equivalency (SE). According to the MFDS regulations, the devices are divided into four categories:
- Recognized SE (Substantial Equivalency) Devices
- SE (Substantial Equivalency) Devices
- Modified Device
- New Devices
Attention
South Korea distinguishes not only classes I to IV, but within class II again the categories 1 to 4.
Devices of categories 1 to 3 require a product certification by the NIDS. For this purpose, an application for product notification must be submitted to the NIDS (analogous to class I devices). This must be supplemented by additional documents.
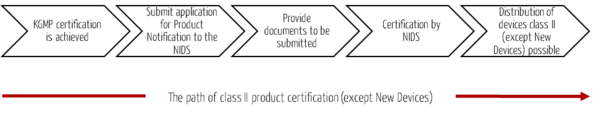
For category 1 devices (Recognized SE Devices), a Test Report of Safety and Performance is required in addition to the application for Product Notification. A processing time of five days and a cost of approximately USD 120 for certification can be expected.
Category 2 (SE Devices) and category 3 (Modified Devices) devices undergo a detailed review of the documents to be submitted in addition to the application for Product Notification. For the review of the additional documents, a processing time of 25 days and costs of approx. USD 120 can be expected.
In the case of category 2 devices (SE Devices), an evaluation of comparison data with devices already on the market must be prepared in addition to the information on category 1 devices.
For category 3 devices (modified devices), documentation of the origin and development process, including the usage status overseas, must be prepared in addition to the information on category 2 devices.
Approval of devices in class II (New Devices) and classes III to IV (4c)
For class II medical devices in category 4 (New Devices) and class III and IV devices, an application for approval is submitted to the MFDS.
There are two options for manufacturers:
- General review of the technical documentation
- Inspection of the safety and effectiveness of the device (SER file)
For the review of the documents, a processing time of 60 to 80 days and costs of between USD 600 and 1,200 (depending on the medical device class) can be expected.

Author:
Florian Krafft

In addition to the approval application and the technical documentation or SER, further relevant information must be provided or submitted:
- All relevant test reports (Validation Documentation)
- Comparison data to devices on the market (Comparison Data)
- Description of the mode of action of the device (Mode of Action)
- Process descriptions of the safety and performance of a device (Safety & Performance Process)
- Reports on clinical investigations (except for devices undergoing SER inspection)
- Information on the status of use abroad
For class IV medical devices, manufacturers must submit the technical documentation in STED format. For classes II (New Devices) and III, submission in STED format is voluntary.
If the technical documentation does not show sufficient equivalence to a device on the market (Comparison Data), the device must undergo a Clinical Data Review (CDR) in addition to the review of the documentation.
Attention
Foreign test reports are only partially accepted for product approval in South Korea. It is, therefore, advisable to have the relevant reports checked in advance with regard to approval.
Summary and conclusion
The South Korean market represents an interesting market for foreign manufacturers due to the annually growing import rate of medical devices. However, there are some hurdles to overcome:
- Foreign manufacturers need a locally based Korean Licence Holder (KLH) regardless of the medical device class.
- The requirements of KGMP (Korean Good Manufacturing Practices) must be met and certified.
- All necessary documents and files must be submitted in Korean.
- Foreign test reports are only partially accepted.
The level of hurdles depends on the class of the device and, for class II devices, the category of the device.
Support with product approval in South Korea
Together with our local partner, we can help you overcome these hurdles. We will be happy to support you in your registration process. Please feel free to contact us.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.