RIMS: Future or anchoring in the past?
Medical device manufacturers have high expectations of Regulatory Information Management Systems (RIMS). The costs and efforts involved are immense and usually much higher than expected. The benefits, on the other hand, are not clear.
This article will give you some hints,
- whether you need a Regulatory Information Management System at all and
- what you should pay attention to when selecting and implementing it.
1. Definition: RIMS
Definition RIMS
A Regulatory Information Management System (RIMS) is a software system that helps pharmaceutical and medical device manufacturers manage regulatory information and comply with regulations. It is a centralized platform that enables the collection, management, and delivery of regulatory data and documents.
Source: Johner Institute
2. Goals of RIMS
Pharmaceutical and medical device manufacturers expect several advantages from the use of a Regulatory Information Management System:
- Higher compliance, minimized regulatory risks
A RIMS is designed to help check documents for completeness and remind of overdue tasks and deadlines. RIMS often also provide an overview of relevant regulatory requirements.
In addition, some RIMS track regulatory changes and provide information about them so that manufacturers can take appropriate action to ensure ongoing compliance.
- Improved efficiency, accelerated approval and time to market
The RIMS is designed to simplify cooperation within the company and with the approval authorities. To this end, it clearly regulates processes and partially automates tasks.
The systems make it easier to search for information and allow all stakeholders to work together on data, from development to the post-market team.
Example
Development must know from the beginning what is required from a regulatory point of view. Regulatory Affairs must know from the outset what is to be developed in order to help with the regulatory strategy. Otherwise, there is a risk that products will not be launched, will not be launched with the planned "features,” or will not be launched in the planned time.
- Improved data quality
The data is available centrally and can be viewed centrally. Both contribute to avoiding or at least quickly correcting erroneous data. Both, in turn, are necessary to make decisions based on reliable information. - Reliable information as a basis for decision-making
The common platform and central processing of data provides companies with an overview, for example, of the progress of approval projects.
This transparency is necessary for well-founded decisions, e.g., about the start of a market launch campaign.
3. Functions of a RIMS
To achieve these goals just mentioned, RIMS offer numerous functions.
a) Management of regulatory requirements
RIMS support the handling of regulatory requirements:
- Central storage of regulatory documents, guidelines, and standards
- Management of regulatory changes and updates
- Follow-up of necessary measures
b) Management of admission procedures
- Follow up on the admission procedures
- Assist in the submission of regulatory applications and documents
- Technical documentation management
- Monitoring of compliance deadlines and tasks
c) Post-Market Surveillance
- Automated collection and filtering of data
- Analysis and trend calculation of this data
- Support in the preparation of reports (e.g., PSUR)
- Follow-up of necessary measures
d) Other
- Reporting and analyses in the area of regulatory information
- Integration with other systems (see next section)
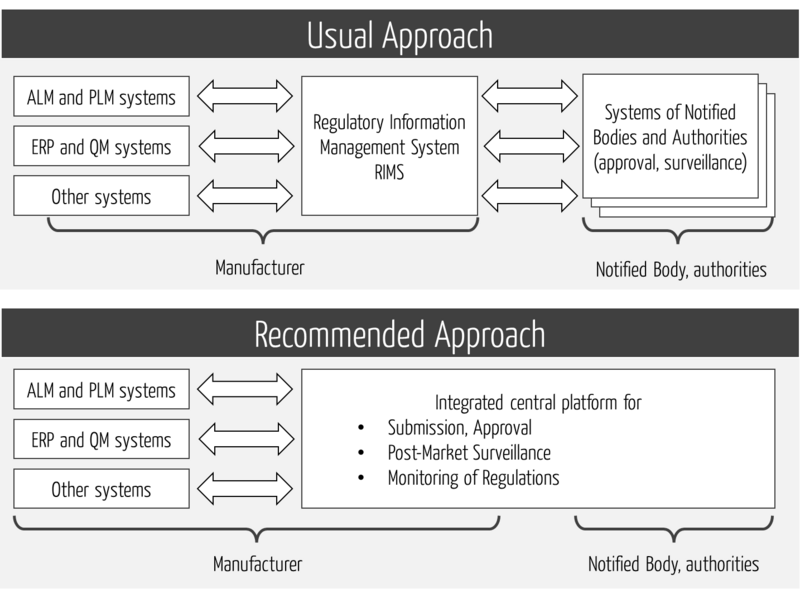
4. Embedding a RIM in the system landscape
a) Overview of the systems
In order to allow the most consistent flow of information possible, regulatory management systems must work together with other information systems.
- Document management systems (DMS) The
integration of RIMS with DMS is particularly relevant. This is the only way to ensure legally compliant storage, control, and versioning of regulatory documents. It must be possible to clearly assign the documents with their respective versions to the regulatory procedures. It is also necessary to have an overview at all times of when which documents were subsequently submitted in which version. - ALM and PLM systems
In the application and product lifecycle management systems, manufacturers keep important data such as the contents of a "design history file," some of which is required for submission. - ERP systems
Enterprise Resource Planning (ERP) systems store relevant product master data. For many manufacturers, the assignment of components and parts to individual medical devices also takes place in the ERP system. It is precisely this data that is relevant for approval procedures and should therefore be able to be seamlessly transferred to the RIMS.
Batch information is essential for product and component traceability and recalls, as well as for compliance with supply chain requirements. - Quality Management Systems (QMS)
Manufacturers also manage records in the QMS, such as the results of verification and validation activities. Manufacturers must also provide this evidence during the approval process. This requires integration of the RIMS with the QMS. - Systems of the authorities and notified bodies
Authorities and Notified Bodies are increasingly demanding that approval documents be submitted digitally. For this purpose, they offer interfaces that a RIMS should support.
There are other systems, such as CAD systems and product information management (PIM) systems. The latter manage product information such as instructions for use, markings, and other labeling. These documents are also part of the technical documentation and must therefore be managed by the RIMS.
b) Information flow
The RIMS thus form the interface to the internal and external systems, for example, at authorities and notified bodies (see Fig. 1).

5. Criteria for the selection of a RIMS
Attention
Typically, companies use feature lists to compare vendors and select the appropriate product. However, such catalogs of criteria are unsuitable for
--- to find out whether a RIMS is the right system at all,
--- assess whether the RIMS is fit for the future, and
--- assess the likelihood that the implementation will be successful.
Therefore, also read the section "Tips for selecting and implementing a RIMS"!
a) Criteria concerning the product
Functionality
The first impulse is to check how completely the system offers the functions mentioned in chapter 2.
This completeness concerns the processes such as:
- Development
- Approval
- Post-Market Surveillance
- Monitoring regulatory changes
The completeness also concerns the markets and their regulatory requirements. A US system that does not recognize the concept of Notified Bodies is of limited use to a manufacturer who wants to market products in the EU.
The system should also support processes specific to its own products. This is because systems that are agnostic (such as a DMS in extreme cases) cannot check product-specific content automatically. This work is then left to regulatory affairs and quality managers.
Example
A system can only verify that all essential performance characteristics of a product have been considered in the "60601-1 tests" if it knows the concept of essential performance.
A system can only check whether all safety-related use scenarios are present in the risk management file if it has modeled these entities and can access them. This is not the case for systems that only manage PDFs and their metadata.
Data-driven instead of document-driven approach
A RIMS should take a data-driven rather than document-driven approach. This is because the algorithms need this data to achieve the hoped-for benefits:
- Automated check of completeness, correctness, and consistency of data for submission, for example
- Automatic analysis of the data, for example, for post-market surveillance
- Through both higher compliance and shorter time to market
Note
A RIMS that "only" enables workflow, management, and versioning of documents is not "fit for the future.”
Interoperability
Chapter 3 shows how many systems a RIMS must work with. Therefore, interoperability with these systems is a basic requirement. Otherwise, another data silo is created.
Attention
Such data silos do not only mean higher effort. They result in redundancies and, thus, usually inconsistencies. As a result, such systems increase regulatory risks instead of reducing them.
The ideal system is one that supports the process as "end-to-end" as possible, minimizing the number of interfaces and thus the problems with interoperability (see Fig. 3).
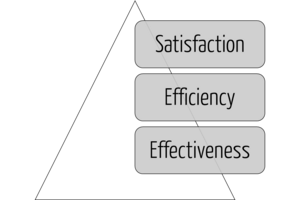
Serviceability
A usable product must first serve the achievement of objectives (effectiveness), then the efficiency of this achievement, and only in the third step the satisfaction of the users (see Fig. 2). Users who do not achieve their goals or do not achieve them efficiently will not be satisfied in the long run.
Attention
Many (potential) users confuse "sexy design" with usability. A modern-looking user interface (UI) equipped with "fancy" dashboards is no substitute for a UI that allows users to complete their tasks quickly and correctly without unnecessary detours (see Fig. 2).
Therefore, the actual future main users should have worked with the system before making a final purchase decision and should be significantly involved in the decision.
Non-functional requirements
The (other) non-functional requirements include:
- The performance and scalability of the systems
- The security and data protection
- Portability, i.e., the ability to be operated on the systems of the (medical device) manufacturer. For web-based systems, the user's browser must be supported by the vendor.
Note
Browser-based systems also require connectors to the manufacturer's systems.
Costs
The costs are mostly composed of:
- License costs (one-time, maintenance costs, subscription) depending on
- Number of users (staggered according to their roles, if applicable)
- activated modules/functionalities
- Usage behavior, such as the number of products or submissions
- Support costs
- Costs for implementation (data migration, parameterization, training)
- Costs for operating the infrastructure (if the system is self-hosted)
- Duration of the commitment
b) Criteria concerning the supplier
Even if a RIMS meets all the checkboxes, it still should not be implemented if the provider does not meet the requirements.
Clear picture of the future and roadmap of the provider
Providers need to have a clear vision of where regulatory systems, medical devices, technologies, and its RIMS are headed.
The provider should have formulated this vision very precisely. It should be congruent with the provider's own vision.
Example
The supplier should clearly outline how long it will continue to consider the submission of technical documentation as (PDF) documents to be timely and at what point it will support document-free submission.
Influence on product design
The providers have to manage a balancing act: On the one hand, they should respond to customer wishes as individually as possible. On the other hand, they must ensure the conceptual integrity of their products and represent their economic interests.
Therefore, medical device manufacturers should only select vendors where they are a typical customer. This is because similar customers (other medical device manufacturers) will place similar requirements on the supplier, which the supplier can then meet more easily.
Attention
Many RIMS providers have their roots and largest customer base in the pharmaceutical sector. Therefore, the influence of medical device manufacturers on product design is mostly limited - even if claimed otherwise during the evaluation phase.
Quality and availability of support
Regulatory Information Management Systems are complex software applications. Therefore, users and people responsible for installation, configuration, and operation need competent and available support.
Attention
It is not enough for support to know how their own product works. They must understand regulatory requirements and be able to explain how they can be met with their system.
Manufacturers should specify,
- in which relevant time zones the support
- at what times is available and
- which response times he promises.
Professional competence of the provider
Rapid technological advancements, particularly in artificial intelligence and hyper-scaling, are having a significant impact on providers:
- Many technical challenges can be solved more easily and quickly. This means that technical competencies remain important in the development of RIMS but are becoming less important.
- In return, the demands on the technical competence of the provider are growing. The provider must be a subject matter expert. In the case of RIMS, this is expertise in regulatory matters (for medical devices!).
For example, a solid half-knowledge is neither sufficient to train systems with artificial intelligence nor to assess their results competently.
Note
Subject matter expertise is also necessary to understand the intricacies of ever-changing regulation and map them into algorithms. Because without an algorithm that automates the review of regulatory requirements, RIMS remain limited to specialized document and task management systems.
This will not enable manufacturers to get their products approved in the shortest possible time or even to move into continuous compliance assessment, as will be necessary for software in the future.
6. The top 10 tips for selecting and implementing a RIMS
Tip 1: Decide if a RIMS is the right approach
A RIMS can solve as many problems as it can create many new problems. Therefore, manufacturers should answer two questions for themselves:
- Is a software system really the solution to the problems?
The introduction of software such as a RIMS results in the manufacturer having one more system if it does not eliminate another in return.
Each additional system causes additional efforts and costs for integration, configuration, and operation.
The benefits must justify these efforts and costs. There are usually root causes for the problems (e.g., regarding compliance, time to market, and efficiency) that a RIMS cannot solve. - If a system appears to be the solution, is a RIMS the appropriate system?
Most RIMS are based on supporting conventional document-based processes. In doing so, you manifest these conventional processes and anchor the manufacturer in the (future) past.
Note
The efficiency and effectiveness of regulatory processes can only be optimized "end-to-end.” Local optimization (e.g., at manufacturers) is of limited help. Therefore, an end-to-end platform is needed (see Fig. 3).
This platform should support all regulatory processes, not just submission. This, in turn, requires that the platform not only integrates the internal systems, but also the external ones (see Fig. 4).
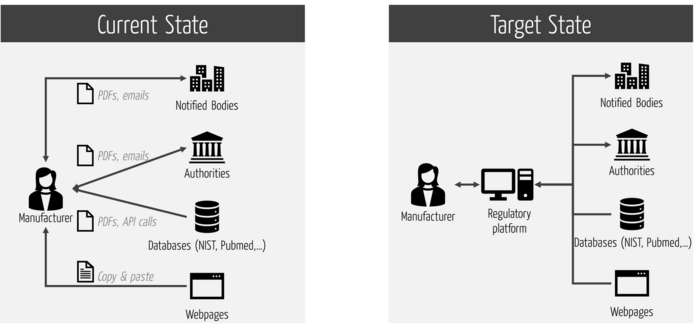
Note
Many RIMS do not support this comprehensive integration. This causes unnecessary interfaces and thus expenses as well as regulatory risks.
Tip 2: Don't be blinded by "feature-itis”
RIMS providers want to impress with an extensive list of features. For example, many providers are currently advertising the artificial intelligence of their products and praising this as proof of the future viability of these systems.
However, artificial intelligence should only be used where it meets actual stakeholder requirements that cannot be met at least equivalently by conventional logic.
Example
One RIMS provider advertises that technical documentation could be created faster with AI (especially Large Language Models). Yet document-based, mostly PDF-based, submission is a concept of the last millennium.
It would be more helpful if the system used conventional logic to check the conformance of the contents of this documentation for completeness, consistency, and correctness, and used AI to transform external unstructured data (e.g., clinical literature) into internal structured data.
The vendors' feature lists are mostly aimed at the decision-making level of the manufacturer. That is understandable. But the decision-makers are usually not the people who will later have to use and operate the system.
Tips
Include all stakeholders in the selection process. This also includes your own IT, which will have to operate the systems later.
Ask other customers of the provider (reference customers) about their personal experiences. Ask specific questions about the duration of the project, what worked and what didn't during implementation. Talk to the real users and ask how the data gets into the RIMS and what post-processing is necessary to submit the data.
Tip 3: Clean up and structure data
The data is distributed across many systems (see above) and files at the manufacturers. For example, they can be found in ALM systems, in Word and Excel files, in e-mails, and user manuals. And some data is missing altogether.
The introduction of a RIMS does not change this yet. But it does force manufacturers to "clean up" this data:
- Correct data
- Complete data
- Eliminate redundancies
- Achieve uniform granularity
- Coding data (e.g., with IMDRF codes)
- Create references between the data
- Eliminate unnecessary data
For this to succeed, manufacturers need
- a clear target image of these data structures,
- Data structures that allow automated checking of data for compliance,
- a process model for this cleanup.
Note
Johner Institute assists manufacturers in this task, which is the mandatory prerequisite for the implementation and success of any RIMS or other system.
Tip 4: Exercise caution with data migration
Many RIMS vendors expect data migration. Vendors should be aware that data migration - as the name implies - means migrating the data from the previous sources into the RIMS. Such a migration poses dangers:
- The data is then available redundantly.
- Data silos are created.
- Data changed in RIMS does not flow back to primary systems such as ALM.
Therefore, manufacturers should
- ensure that there are no redundancies,
- determine which is the leading system and
- ensure that data is not lost that is unimportant to the RIMS but relevant for other purposes.
Tip 5: Perform trial operation
Only a trial run shows,
- the extent to which the RIMS meets the specific requirements of the manufacturer,
- how flexibly the supplier responds to the manufacturer's wishes,
- what effort is actually required to connect to the company's own systems (for example, for the development of connectors),
- how competent the support is and
- how long the project will take.
Note
A product demo is only enough to narrow down the list of vendors. But not to answer the above questions.
The manufacturer should also plan for trial operation. These include:
- Questions to be answered by the trial operation (resulting in own products, markets, own organizational units to be considered in the trial operation)
- Success and termination criteria (communicate these to the provider).
- Own financial and human resources (which are almost always scarce)
- Project organization (meetings, steering committee, etc.)
Tip 6: Do not choose a big bang rollout
Even with a successful trial, big-bang rollouts are not a recommendation. The organizational and technical changes are too extensive for the organization to digest. The overall project can be broken down according to various aspects:
- Processes to be supported
- Affected organizational units
- Products or product classes
- Markets (e.g., EU, USA)
- External data sources and organizations
Tip 7: Take change management seriously
It sounds banal, but nevertheless often comes up short due to time and cost pressures:
Careful planning and communication are critical to ensure that all affected employees understand and accept the implementation of the RIMS. Training and education materials should be provided to familiarize users with the new software.
Tip 8: Meet QM requirements
Every new system attracts the interest of auditors. They ask not only about the competencies and training certificates but also about the revised instructions (SOPs, work instructions, and their approvals) and the validation of the system.
Further information
This article on Computer Systems Validation shows how to validate software such as a RIMS and which standards help.
The effort and timing for validation must be included in the project plan.
Tip 9: Establish internal "evangelists”
The likelihood is high that the initial enthusiasm will evaporate when the first bugs and the whole effort for the project become visible.
Therefore, it is useful if internal employees can act as competent evangelists to
- move the project forward,
- coordinate the requests for further development and adaptation of the software,
- form the interface to the supplier and
- serve as a readily available point of contact for questions and difficulties.
Tip 10: Describe and calculate exit scenario
According to Standish Group, most IT projects fail to meet goals on time and within budget, or they fail altogether. Therefore, it is part of risk management to plan for this failure:
- Ensure exit clauses in contracts
- Calculate exit costs
- Describe exit scenarios, such as how the company will "migrate back" or continue to maintain processes without the RIMS or its vendor
7. Conclusion and summary
a) Systems are not yet a solution
No manufacturer needs a RIMS. But every manufacturer needs effective and efficient regulatory processes to
- to bring its products quickly and safely to the markets,
- continuously ensure the conformity of these products and his company, and
- to make do with limited resources (especially regulatory affairs experts).
Regulatory Information Management Systems (RIMS) can be a solution to this, but they are not always.
A RIMS can even create new problems:
- Additional interfaces and increased complexity of systems and processes
- Financial and regulatory risks associated with the implementation of the RIMS
- Anchored in the past by a RIMS that cements old concepts like document-based submissions
Many RIMS providers "only" want to sell software. However, digital transformation does not succeed through software implementation alone.
Further information
In this article on digital transformation, you will learn what needs to be transformed and how this transformation is most likely to succeed.
b) Requirements must be met
This digital transformation will only succeed through
- a precise target picture of future regulatory systems and technical solutions,
- a clear roadmap to achieve these goals and to be compatible with future regulatory systems,
- a regulatory expertise on the part of the manufacturer and the provider to implement this roadmap (for example, to automate the review of technical documentation) and thus actually achieve the expected benefits.
In particular, RIMS providers that have a focus on the pharmaceutical environment often do not have this deep expertise that medical device manufacturers need.
Note
The Johner Institute has a precise target picture and helps companies with the digital transformation with consulting and software solutions.
Schedule a free consultation to avoid unnecessary risks when implementing systems and ensure compliance with transparent, streamlined, and automated regulatory processes to bring your own products to market quickly and safely.

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.