Classification and registration pathways of medical devices and IVDs in the U.S.
Medical devices are classified based on risk and assigned to Class I (low risk) to III (high risk). This classification applies to both medical devices and IVDs, and the classification procedure is also identical. Classification rules known from Europe do not exist at the FDA, and the FDA determines the classification for generic product types.
Classification is one part of determining the registration path. However, the registration path is not determined solely based on the classification but based on the intended purpose and related requirements (controls) to ensure the safety and effectiveness of the product.
"Controls": Requirements for medical devices
All medical devices in the USA are subject to the so-called "General Controls." These describe the basic requirements of the FDA and include:
- Adulteration;
- Misbranding;
- Device registration and listing;
- Premarket notification;
- Banned devices;
- Notification, including repair, replacement, or refund;
- Records and reports;
- Restricted devices; and
- Good manufacturing practices.
Class I products are subject only to the "General Controls." These are sufficient to ensure the safety and effectiveness of the product. For Class II and III products, the FDA has defined further requirements and the General Controls to ensure safety and effectiveness.
As a rule of thumb, one can state:
- Class I and II products are cleared through the 510(k) process - unless exemptions are defined for the product code in question. For example, under ¾ of Class I, products are exempt from premarket notification (510(k)) requirements. Some products are exempt from specific Good Manufacturing Practices requirements (Quality System Regulations - 21 CFR Part 820).
- Class III products are registered via the PMA process - unless exceptions are defined for the relevant Product Code.
The following table lists the most common approval pathways based on product risk:
Table 2: Registration paths depending on the requirements
Class | Controls | Exemptions | Registration path |
Class I | General Controls | With Exemptions | Listing |
Without Exemptions | 510(k) | ||
Class II | General Controls and Special Controls | With Exemptions | Listing |
Without Exemptions | 510(k) | ||
Class III | General Controls and Premarket Approval (PMA) | / | PMA |
Now how do you determine the classification and registration process?
The FDA determines the registration process. As described above, this is done based on the intended use and the necessary controls. For this purpose, the FDA maintains a database with superordinate product groups (panels).
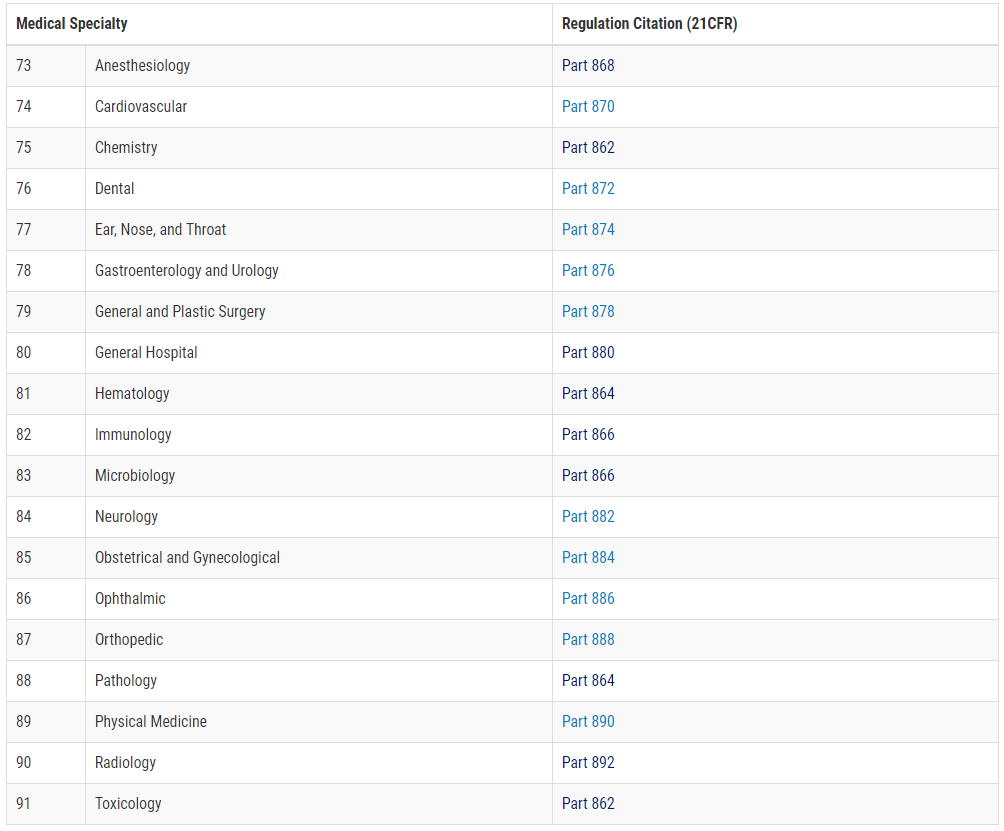
Figure 1: Classification Panels; Source: FDA
Note: In Vitro Diagnostics can be found in the Chemistry (21 CFR 862), Hematology (21 CFR 864), and Immunology (21 CFR 866) panels.
Within the panels, the FDA defines product types based on a general product description, for each of which a Regulation Number is defined. The FDA assigns product codes for specific product types within the Regulation Numbers.
There are now more than 1700 definitions for different types of products in the legislation.
For each product code, the FDA defines
- The Class
- The registration procedure
- One Regulation Number
- A product code
- Requirements for the QMS
- As far as applicable:
- Applicable Special Controls
- Further descriptions of the product (e.g., technical information or area of application)
If you want to determine the classification and the registration path, you need the correct product code. The following graphic shows how you can proceed:
What to do when...
The assignment to a product code is not always clear. What can you do if
1. There is no matching product code?
In this case, your product is automatically classified as a Class III product and would need to be approved through the PMA process. However, suppose it is a product that the FDA would likely classify as Class I or Class II because General Controls or General and Special Controls are sufficient to ensure the safety and efficacy of the product. In that case, a De Novo process can be followed.
This is a procedure for novel Class I and Class II products for which there is no comparator product on the market.
2. More than one product code is applicable?
If you find several product codes that may apply to your product, you should check under which product code comparable products are registered. To do this, you can use the Registration and Listing Database, for example, and search for competing products.
3. You are unsure whether you have determined the correct product code?
The first step here is to check the intended use of other products under the Product Code you have identified. If you are still unsure, you can ask FDA for a formal evaluation of your product and classification under a Product Code. The process required to do this is called a 513(g) Request. FDA has issued a guidance document describing the process, "FDA and Industry Procedures for Section 513(g) Requests for Information under the Federal Food, Drug, and Cosmetic Act Guidance (2012)."

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.