MDR - Distributor Requirements (that also affect the manufacturers)
Tuesday, October 15, 2019
The medical device regulations have significantly increased the requirements for distributors. You need to understand these requirements to avoid multi-year custodial sentences threatened in the event of infringement.
These distributor requirements also (indirectly effect) the manufacturers(!). Therefore these tips are helpful for manufacturers to take the hard line with the distributors. This creates a higher legal security and better control for the manufacturers about the sales channels.
Content Overview |
Find out about a helpful guideline from the Irish authorities in the information below.
1. What a “distributor” is (and what it isn't)
a) Definition
The Medical Device Regulations (MDR) has many roles, under which the economic operators are listed. These economic operators include the distributors as well as the manufacturers, the EU authorities and the importers (see figure 1).
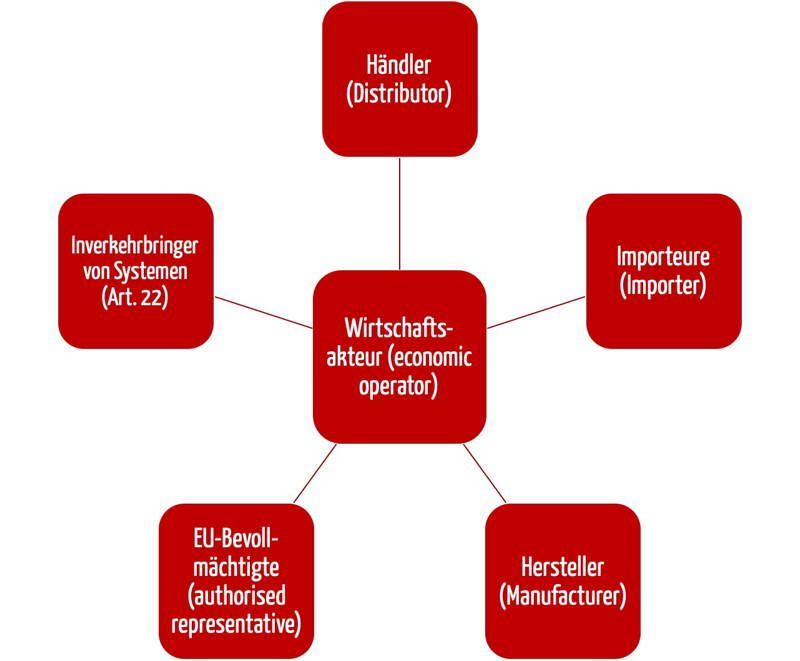
Definition: Distributors
“Distributors” are any natural or legal person in the delivery chain, who provide a product on the market up to the time of commissioning, with the exception of the manufacturer or importer.
Source: MDR, Article 2
b) Typical activities
The typical activities of a distributor include:
- Purchase of products from manufacturers or other (interim) distributors
- Marketing and sale of products to end-customers or other distributors
- Storage and transport of products
- Possibly attaching own labels (be careful here! More on this later).
- Instruction of users
- Support in installation and commissioning
- Answering user questions
- Handling customer complaints, feedback to manufacturer
Some distributors also deal with the service and repair of products or organize these.
c) Differentiation of the role of distributor from the importer
If a distributor purchases the products from a manufacturer or another distributor that is not based in the EU, this distributor also assumes the role of the importer. The MDR defines importers as follows:
Definition: Importer
“An “importer” is any natural or legal person based in the Union who markets a product from a third-party country on the Union market;”
Source: MDR, Article 2
The MDR imposes additional requirements on importers.
d) Differentiation of the role of distributor from the manufacturer
Many distributors may sell the products under their own name and not name the actual manufacturer. Thus the distributor becomes the manufacturer. The Medical Device Regulations defines manufacturers as follows:
Definition: Manufacturer
“Manufacturers” are a natural or legal person that produces a product or refurbishes or develops one or has one developed, refurbished or manufactured and markets this product under its own name or own brand;”
Source: MDR, Article 2
With the MDR, the PLM-OEM constructs are also a thing of the past. For this, the MDR, under article 2 (16) (2) offers distributor new options: It no longer sees the following activities as a product change that has an effect on the conformity of the product:
- Provision of information for a product. This also applies to translations.
- Changes to the external packaging
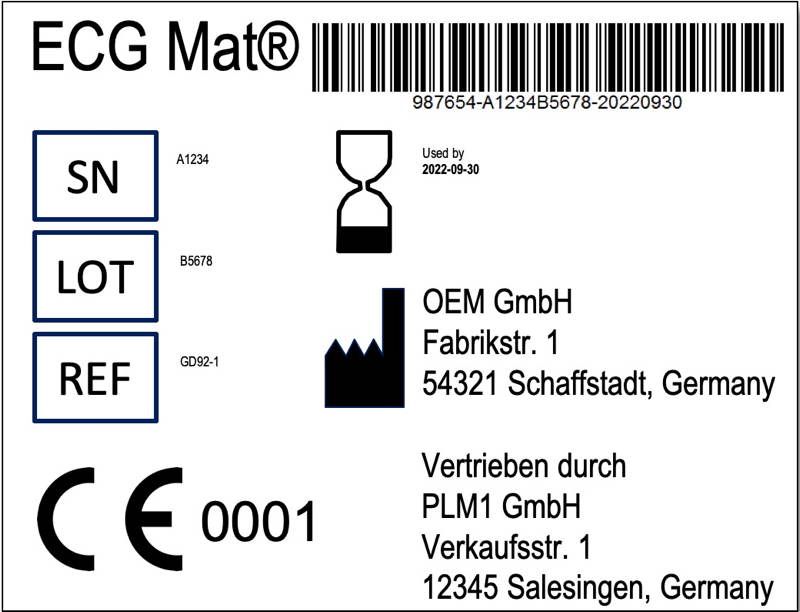
Naturally, it is also possible that the manufacturer markets the product in the distributor's “design”. The manufacturer must, however, always (also) be visible on the label. This is required by Article 16, paragraph 3.
2. The obligations that distributors must fulfil
a) Background / History
The requirements set for the distributor by the Medical Device Regulations MDR are derived from a superordinate framework for marketing of products. This is also called the “Goods Package” and is based on:
- EU directive 765/2008 “on the regulations for accreditation and market surveillance within the context of marketing of products”
- Resolution no. 768/2008/EU “on a common legal framework for the marketing of products”
b) Obligations of the distributors as a testing authority in the delivery chain (from the viewpoint of the MDR)
The thought of the MDR is that every economic operator ensures, if possible, that the economic operator has fulfilled the regulatory requirements one step previous in the delivery chain.

The reviews required by the MDR for distributors include:
- Does the product bear a CE symbol?
- Has a declaration of conformity been issued for the product?
- Has the importer listed its name and address on the product, packaging or a document.
- Has the importer not covered the labels of the manufacturer with its own additional(!) labels?
- Has the manufacturer issued a UDI?
- Does the product appear to conform to the legal requirements?
If one of these conditions is not fulfilled, the distributor may not sell the product and must inform the manufacturer, importer and the EU representatives.
Caution!
Distributors may no longer blindly relay on the manufacturer and importers. They must exactly review whether the manufacturer has valid certificates, in particular in the transition period.
c) Obligations of the distributors as a testing authority in the delivery chain (from the viewpoint of the MDG)
The MDR allows distributors to perform random testing in Article 14 paragraph 2. They should be able to review whether the products actually have a CE symbol, a declaration of conformity and a UDI.
The Medical Device Implementation Law MDG appears to be more rigid: There is a threat of a custodial sentence under article 59(2) if someone offers falsified products, stores them or sells them. This affects the distributors directly.
In order to avoid this type of infringement, and thus a custodial sentence, every distributor must perform a 100% goods receiving inspection of the goods. So one may fear that the MDG annuls the random sample inspection.
It even gets more absurd:
If the distributor discovers a falsified product in the warehouse, they are liable. However, according to article 7 MDG, it cannot be sent back, because then it falls “out of the scope of validity of this law”. This is then also punishable.
And the distributor can also not destroy it. Because then they are also culpable, because they must “provide free samples of the product at the request of the authorities”, or, “if this is not practical, allow access to the product” (MDR Article 14(6)).
A reader who brought our attention to this conflict (thank you for that), writes:
“I'm curious about the assistance of the legislators about how these contradictions should be solved by a distributor, e.g. by Aldi, which offers medical devices once or twice a year (e.g. blood pressure monitors) within the framework of any special offers.”
d) Requirements for the activities of the distributors
The MDR obligates the distributors to additional activities:
- Storage and transportation pursuant to manufacturer specifications
- Collecting complaints and incident reports and forwarding them to manufacturers and possibly importers. This also applies to products for which the distributor itself has doubts about the conformity.
- Keeping a “register” of non-conforming products, recalls and withdrawals
- Informing authorities (in Germany the BfArM pursuant to article 44 of the MDG) about unsafe and falsified products and about corrective actions and provide information and documentation upon request.
Caution!
With the MDR, distributors and importers become part of the post-market surveillance and reporting system. They must actively cooperate in this, which requires that they be able to trace the products. This is explicitly described in the MDR in article 25 and by the EU in its “Factsheet for Authorized Representatives, Importers and Distributors”.
e) Registration of distributors
In Article 30, the MDR mentions the “Electronic System for the Registration of Economic Operators”. In it, it only obligates the manufacturers, authorized representatives and distributors. The MDR leaves it to the member states to issue conditions for the registration of distributors.
And that's just what member states such as Germany appear to be doing: In the Medical Devices Implementation Act MDG it states:
The Federal Ministry of Health is empowered to require through a statutory regulation, that distributors providing products on the German market report this to the responsible authorities before starting their activities. In the statutory regulation under section 1, the content and form of the notification may also be proscribed.
MDG §55(10)
Thus a national ordinance is to be expected, that describes this obligation to register.
f) Special cases for the quality management system
As described above, manufacturers may repackage products and provide and translate the legally required information (Annex I, section 23). Thus resulting not only in the previously mentioned obligation of indicating the name of the distributor on the label (see fig. 2). Much more, the distributors must have a quality management system.
This QM system must regulate all relevant actions:
- Repackaging of the product (and checking that this repackaging does not affect the conformity)
- Creation / provision of information (and checking that these fulfil the legal requirements)
- Translating the information
- Receive / order (?) information about the manufacturer's corrective actions
There is no obligation that the distributors have their QM system certified, e.g. pursuant to ISO 13485. This is also confirmed by the Irish authorities. However, these authorities provide for additional activities as part of the QM system:
- Personnel, Training
- Managing documents and diagrams
- Receipt, storage and provision of medical devices
- Handling returned products
- Handling falsified products
- Recall of products
- Contracted processes
- Transport
- Audits
- Supplier management
- Management Review
- CAPA
- Waste management
- Internal and external audits
- Validation including Computerized Systems Validation
- Etc., etc.
Caution!
Note, that as a
- Distributor: the authorities require that you implement a complete QM system comparable to ISO 13485.
- Manufacturer: in the case of article 16(4), you must certify to the distributor that its QM system meets the requirements of just this article.
So that the manufacturers can test the QM system, the MDR requires the distributors to notify “its” manufacturers and the responsible authorities(!) 28 days before the provision of the product. The MDR mentions “notification”, not “asking for approval”.
g) Obligations not(?) imposed on the distributors by the MDR
The distributors are not obligated to register the products. This is the task of the manufacturers or importers. The distributors also do not require “Person Responsible for Regulatory Compliance”.
It is unclear whether the distributor is responsible for the storage and transport if the manufacturer delivers directly to the distributor's customers. The question of whether the importer or the distributor is responsible for the transport from the importer to them, the distributor also leads to discussion.
3. What the manufacturer must take into account regarding their distributors
a) Problem
Some manufacturers would like to think that all of these regulatory requirements are the distributors’ problems, and not their own. But that's not the case:
Distributors may even “relabel” products and “repackage” them, e.g. in their own design. They must only “notify” the manufacturer, not ask for approval.
This means that the manufacturers can't (just) unilaterally inhibit these activities. On the other hand, they are obligated to perform Post-Market Surveillance. This is difficult if distributors sell products in other countries unknown to the manufacturer with translated support materials.
b) Post-Market Surveillance
Manufacturers should proactively search around the world for information on their own products. Not only in the countries where they know that their products are being sold. Automation such as Post-Market Radar of Johner Institute helps in this.
It is essential that the manufacturers, together with their distributors, authorized representatives and importers, exactly determine who is responsible for which activities (not only) within the framework of the Post-Market Surveillance.
Naturally, this surveillance should also focus on falsified products and unauthorized distributors even after the product is on the market.
c) Setting the hard line for distributors
Manufacturers should monitor their distributors just like the products themselves.
In the case of distributors that “repackage” or “relabel” products, the manufacturer must also certify the conformity of their QM systems. This requires that the manufacturer has checked this.
Tip!
Manufacturers can use just this test to sort out unsuitable distributors.
Sometimes it is helpful to select the proper use and design of the product (e.g. plugs) so that certain markets are excluded. Thus manufacturers can somewhat dam the “misuse” by undesirable distributors.
In addition, the manufacturers should enter into quality assurance agreements with the distributors. A QAA should include, in particular:
- Affected products or product groups
- Duration of validity of the agreement
- Contacts for both parties
- Form and deadline for feedback from the distributors to the manufacturer
- The right of the manufacturer to audit / inspect the distributor
- Obligation of the distributor to report to authorities
- Obligation of the distributor to trace products ("register”)
- If necessary, prohibition on sale to other distributors
- Obligation of the distributor to inform importers and EU representatives
- Keeping test samples available
- Obligation to provide support materials
- Obligation to only work with a certified translation agency
c) Caution with Fulfillment Partners
Certain logistics partners offers services far beyond that of a regular package service (like DHL). These so-called “Fulfillment Centers” or (“Fulfillment Houses”) store products, package them upon request, take over invoicing, etc.
Then we aren't talking about "neutral service providers” any more, but distributors in terms of the MDR.
Our tip: Use the specifications of the Blue Guide of the EU.
4. Conclusion / Summary
The MDR has significantly increased the requirements for distributors. This is comprehensible, because only all of the entities involved in the logistics chain can, together, trace where these products are located and whether they are compliant.
Only together can the economic operators react to non-conformities and increase patient safety. So the MDR removes the possibility of finger-pointing.
The distributors should have a (comprehensive) complete quality management system.
The manufacturers should know that the obligations of the distributor have at least an indirect effect on them.
So we hope that the MDG does not lead to contradictions with the MDR, and that a higher safety of the patients and not an increase in bureaucracy are not the results of these legal intensification.
The Irish authorities have published a guideline for distributors with additional practice tips.

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.