EU HTA Regulation 2021/2282 – What the Harmonized Health Technology Assessment Will Bring
In Regulation 2021/2282, the EU has created an instrument for standardizing health technology assessments (HTAs) across the Union. The aim is to make it easier for member states to assess the effects of new health technologies and their pricing on health policies. In an ideal world, this will mean manufacturers no longer have to duplicate their work. However, the EU legislator has made some compromises in the HTA Regulation that could lessen its effect.
Find out what the most important parts of the HTA Regulation are.
1. HTAs: The basics
a) Definition
Definition: Health technology assessment – HTA
Health technology assessment (HTA) is a scientific evidence-based process that allows competent authorities to determine the relative effectiveness of new or existing health technologies.
Source: HTA Regulation, Recital 2
The HTA focuses on comparing one health technology (e.g., medicinal products, medical devices, in vitro diagnostic medical devices, medical procedures, disease prevention measures, diagnostic and treatment procedures) with other technologies. This can allow, for example, the efficacy, safety, and economic, social and ethical aspects of health technologies to be better assessed. That’s why the HTA includes both clinical (e.g., efficacy, safety) and non-clinical (e.g., economical, ethical, organizational, social and legal) aspects.
b) Benefits for decision makers
In practice, HTAs are principally used by states as a basis for health policy decisions, for example, decisions on the allocation of budgetary resources in the health sector. HTAs are also key to deciding which healthcare technologies should be reimbursable. In Germany, corresponding regulations can be found primarily in SGB V.
However, HTAs are also relevant to other healthcare stakeholders: for example, clinics and healthcare professionals can use HTAs to help them decide which technology to use in a therapy. Therefore, HTAs can also indirectly improve patient health by helping improve access to new and/or improved technologies.
c) Benefit for manufacturers and developers
For manufacturers, HTAs are relevant because they influence decisions on the reimbursability of their technology. Reimbursement, in turn, affects the level of demand from hospitals and health centers for a technology.
Manufacturers can also benefit from HTAs when preparing their clinical evaluations. This is because, for the relevant/equivalent devices, the HTAs contain a:
- Carefully compiled literature search and literature review
- Description of the state of the art
- List of the benefits and risks
- Evaluation of the benefit-risk ratio
Manufacturers have to take all these aspects into account in the clinical evaluation and can take them in whole or in part from the HTAs. This doesn’t just save work. It also reassures manufacturers and notified bodies that the clinical evaluation is accurate and complete.
2. General information on Regulation 2021/2282
a) Aim
Up to now, HTA procedures in the EU have been carried out at a national level. Unfortunately, this can result in different member states asking health technology developers for very different data at the same time. This multiplies the time and effort developers have to put in and, in some cases, they even have to submit the same documents twice. In addition, the outcome of national HTAs can vary. So, a technology may be reimbursed in one country, but not in the next.
As a result, the new HTA Regulation 2021/2282 on “health technology assessment and amending Directive 2011/24/EU” harmonizes the process at an EU level. It promises to reduce the burden on companies by requiring much of the documentation to be submitted only once at the EU level instead of multiple times to different member states.
Its aims include unifying HTA methodology and providing an equal basis for decision-making for member states across the entire EU (e.g. with regard to reimbursement eligibility) through joint clinical assessments of health technologies such as pharmaceuticals, medical devices and in vitro diagnostic medical devices.
The main objectives of the HTA Regulation include:
- Uniform rules for HTAs
- An EU-funded foundation for common procedures
- Central coordination
- A platform for cooperation
The Regulation aims to achieve a high level of health protection for patients and users while ensuring the smooth functioning of the internal market with regard to medicinal products, medical devices and in vitro diagnostic medical devices.
b) Scope
The HTA Regulation focuses on the clinical aspects of HTAs, i.e., the relative clinical effectiveness and relative clinical safety of a new health technology compared to existing technologies. The EU HTA is not just limited to the clinical part of the HTA, it is also intended to be absolutely objective. The classification and evaluation of the findings made at the EU level are left to the member states.
In fact, health technology developers will have to deal with two HTA systems in the future: the EU’s system and the national one.
- EU HTA: Provides a report on objective clinical aspects of the HTA
- National HTA: Uses the EU HTA as a basis and incorporates additional aspects (such as economics and ethics). The evaluation of the two parts of the HTA is the responsibility of national bodies.
Member states’ HTA bodies will also conduct joint scientific consultations to advise technology developers on clinical study design to ensure they are able to provide appropriate evidence.
“Horizon scanning” exercises will identify, at an early stage, promising health technologies. In addition, voluntary cooperation at the member state level will be encouraged.
Specifically, the HTA Regulation features:
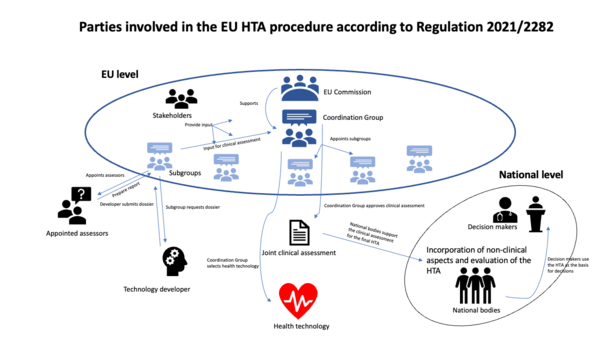
- Coordination Group (Art. 3-6)
The core of the HTA Regulation is the establishment of a Coordination Group to oversee the overall coordination of HTAs at the European level and the cooperation of states, companies and associations.
- Joint clinical assessments (Art. 7–15)
The Coordination Group or its subgroups appoint assessors who carry out joint clinical assessments of health technologies. Manufacturers must submit a dossier for this assessment. It can then be used, for example, by EU member states as a basis for their health policy decisions.
- Joint scientific consultations (Art. 16–21)
If a joint clinical assessment has not been planned yet for a Health technology, its developers can request a joint scientific consultation. The joint scientific consultation generates an evidence base that can be used as the basis for a subsequent joint clinical assessment.
- Emerging health technologies (Art. 22)
The Coordination Group will monitor emerging health technologies and decide which ones will be included in a joint clinical assessment.
- Voluntary cooperation (Art. 23)
The Commission also wants to encourage cooperation on new health technologies between member states beyond the joint clinical assessments and joint scientific consultations.
- Stakeholder network (Art. 29)
A stakeholder network that can provide inputs for the process will be established. An official call for applications to be part of this stakeholder network addressed to all eligible stakeholder organizations will be published.
- IT platform (Art. 30)
All information exchanges will take place via a newly created IT platform.
c) Deadlines
The new HTA Regulation 2021/2282 came into force in January 2022 and will apply from January 12, 2025, followed by a phased roll-out lasting until 2028.
“In order to ensure the smooth establishment and operation of Union-level joint clinical assessments, as well as to safeguard their quality, it is appropriate to start with a small number of joint clinical assessments. As from three years after the date of application of this Regulation, a progressive expansion of the number of joint clinical assessments should take place.”
HTA Regulation, Recital 53
Priority is given to medicinal products, or more specifically, medicinal products for the treatment of cancer. It is true that, from 2025, certain medical devices and IVDs will be selected for a joint clinical assessment “at least every two years”. Nevertheless, in view of the aforementioned priority given to medicinal products, it is likely that no or very few developers of new technologies in the medical technology and IVD sectors will be affected in 2025.
The deferral of the start of application by three years is intended to ensure that there is sufficient time to build the organizational framework required by the HTA Regulation (e.g., the Coordination Group and its subgroups, the stakeholder network). In addition, large parts of the EU HTA process are based on delegated legal acts and guidelines that have still not been passed by the responsible bodies.
The first meeting of the Coordination Group is provisionally scheduled for June 2022.
d) Background
Even before this Regulation, attempts had been made to coordinate HTAs at the European level via the EU co-funded European Network for Health Technology Assessment (EUnetHTA). However, these attempts were all voluntary and not very effective. As the EU Regulation is directly binding in member states, it is expected to be more effective.
3. Content of Regulation (EU) 2021/2282
a) Coordination Group (Art 3–6)
One of the key elements of the HTA Regulation is the establishment of a Coordination Group. It will work closely with the EU Commission. Although the HTA Regulation will not apply until 2025, the first meetings of the Coordination Group are scheduled to take place as early as June 2022.
The Coordination Group will have the following structure:
- The EU member states appoint the members
- Each state has one vote
- The members of the Coordination Group will appoint their national or regional subgroups, which will include HTA experts
Duties
The main duties of the Coordination Group include:
- Coordination of the subgroups
- Identifying emerging health technologies
- Creating strategic and methodological guidance documents for national subgroups
- Specification of procedural steps and timeframes for joint clinical assessments, updates of joint clinical assessments and joint scientific consultations
- Involving stakeholder organizations
b) Joint clinical assessments (Art. 7–15)
Another key aspect of the HTA Regulation is the joint clinical assessment (JCA). However, these assessments are limited to the description of the health technology and the purely factual review of its technical and clinical characteristics. The member states are still responsible for the assessment of all non-clinical aspects and the corresponding conclusions, such as reimbursability.
Scope
According to Art. 7 of HTA Regulation, from 2025 the Commission will select medical devices and in vitro diagnostic medical devices for a joint clinical assessment every two years. However, this only applies to:
- Class IIb or III medical devices
according to Article 51 MDR (Regulation (EU) 2017/745) for which the relevant expert panels have provided a scientific opinion in the context of the clinical evaluation consultation according to Article 54 MDR.
- Class D in vitro diagnostic medical devices
according to Article 47 IVDR (Regulation (EU) 2017/746), for which the relevant expert panels have provided their views in the context of the procedure according to Article 48(6) IVDR.
However, medical devices and IVDs will generally not be subject to an HTA until they have been launched on the market:
“The establishment of a timeframe for the joint clinical assessments for medical devices and in vitro diagnostic medical devices should take into account the highly decentralised market access pathway for those devices and the availability of appropriate evidence data required to carry out a joint clinical assessment. As the required evidence may only become available after the medical device or the in vitro diagnostic medical device has been placed on the market, and in order to allow for their selection for joint clinical assessment at an appropriate time, it should be possible for assessments of such devices to take place after their placing on the market.”
HTA Regulation, Recital 37
The criteria for the selection of medical devices and IVDs for an EU HTA include:
- Unmet medical needs
- Novelty (first in class)
- Potential impact on patients, public health or healthcare systems
- Incorporation of artificial intelligence, machine learning technologies or algorithms
- Significant international dimension
- Major EU-wide added value (such as an increase in population health, economic added value, etc.)
Process
A joint clinical assessment contains the following steps:
- Initiation by the subgroup (Art. 8)
The subgroups initiate joint clinical assessments by appointing a suitable assessor and co-assessor from different member states.
- The subgroup establishes the scope of the assessment (Art. 8)
The scope of the assessment must include all relevant parameters for the assessment with regard to:- The patient population
- The intervention or interventions
- The comparator or comparators
- The health outcomes
- Information provided by the health technology developer
- Inputs received from patients, clinical experts and other relevant experts
- Member states’ interests
- Health technology developer submits a dossier (Art. 10, 11)
The Commission requires the developer to compile and submit a dossier containing all the necessary information on the technology.
The content is further defined by Annex II of the HTA Regulation and delegated acts.
A company may not submit evidence at a national level that is already available at an EU level.
If new clinical data becomes available during the assessment process, the health technology developer concerned must proactively inform the Coordination Group (Art. 11).
- Performance of the assessment
The assessor and co-assessor assess the technology based on the dossier and the defined assessment scope.
The assessor and co-assessor may also consult databases and other sources of clinical information, such as patient registries. - Preparation of the report
The assessor and co-assessor create an analytical assessment report and a summary report (JCA report; Art. 11(1)).
According to Article 11, the draft reports for medical devices and IVDs will be endorsed by the Coordination Group in compliance with the timeframes established in Article 3(7)(e) and Article 15(1)(b). These articles essentially state that the Coordination Group establishes the timeframes itself.
- Comments from subgroup members, the manufacturer and others are incorporated
The revised draft is sent to the Coordination Group. - Finalization of the joint clinical assessments (Art. 12)
- The report is published on the IT platform established by the Commission for this purpose.
c) Joint scientific consultations (Art. 16–21)
Joint scientific consultations come before joint clinical assessments. Health technologies for which a joint clinical assessment has still not been planned but which are likely to be the subject of a joint clinical assessment are eligible for joint scientific consultations.
So, unlike the joint clinical assessment, the joint scientific consultation is conducted:
- If the technology has not yet been selected by the Coordination Group
- but it is promising and
- at the request of the developer.
Therefore, the scientific consultation is of particular interest for developers who want to bring their technology to the attention of the Coordination Group.
Joint scientific consultation on request
Health technology developers can request a joint scientific consultation. The developer's request has to be accepted or rejected within 15 business days following the end of the request period.
Selection criteria include:
- Unmet medical needs
- First in class product
- Potential impact on patients; public health or healthcare systems
- Significant cross-border dimension
- Major Union-wide added value
- Union clinical research priorities
Benefits of joint scientific consultations
According to Article 16 of the HTA Regulation, the Coordination Group conducts a joint scientific consultation in order to exchange information with developers about their plans for a specific health technology. These consultations are also intended to make it easier for developers to later generate the evidence required for the joint clinical assessment.
The joint scientific consultation includes a meeting with the developer and ends with an outcome document that outlines the scientific recommendation made.
d) Emerging health technologies (Art. 22)
The Coordination Group doesn’t just track the development of new technologies on request. It will also write regular reports on emerging health technologies based on existing scientific reports or initiatives on emerging health technologies and information from relevant sources (clinical study registers and scientific reports, European Medicines Agency (EMA) documents – the EMA is now taking on roles in medical device development – etc.)
e) Voluntary cooperation (Art. 23)
The Commission also wants to support cooperation between member states on new health technologies and the exchange of scientific information beyond the joint clinical assessments and joint scientific consultations. The Coordination Group is also expected to encourage such exchanges.
f) Stakeholder network (Art. 29)
The Commission will establish a stakeholder network. The stakeholder network will support the work of the Coordination Group and its subgroups when requested.
How the network will be created
An open call for applications to the stakeholder network will be sent out to all eligible stakeholder associations, in particular:
- Patient associations, consumer and non-governmental organizations in the field of health
- Health technology developers
- Health professionals
The selection criteria will be set out in the open call for applications and will include:
- Proof of current or planned engagement in HTA development
- Professional expertise relevant to the stakeholder network
- Geographical coverage of several member states
- Communication and dissemination capabilities
g) IT platform (Art. 30)
The HTA Regulation is planning another IT platform to be used for all communication. According to Art. 30, this platform will have two main parts:
- A website on which the reports and additional information will be published
- A platform for the secure exchange of information. This platform will be used, for example, for the exchange of data between the Coordination Group and its subgroups, between the member states and the Coordination Group, or for the exchange of data with developers.
4. Effect of the HTA Regulation on manufacturers
a) What are the obligations?
First and foremost, manufacturers who have developed a new health technology must provide documentation on their technology on request. Exactly which documents will be requested has still not been specified.
This request from the Coordination Group for the submission of documents will include a deadline as well as more detailed information on the form and scope of the documents to be submitted.
b) What happens if developers do not meet the obligations?
If a developer does not comply with the request or doesn’t do so completely, they will first receive a second request. If the developer also fails to comply with this deadline, the Coordination Group will end the HTA for the technology and will publish a corresponding official statement on the IT platform.
However, the Coordination Group may re-start the HTA if the developer has submitted corresponding documentation at the national level.
Fines etc. are not currently provided for.
c) What benefits does the new EU HTA regulation promise?
First and foremost, the HTA Regulation standardizes the rules for HTAs across Europe. As a result, the Regulation potentially benefits patients who can rely on a more uniform level of assessment in all member states. This standardization, the resulting more transparent rules and the comparable assessments in the individual member states could also make the work of policy and healthcare decision-makers easier.
Advantages for companies
In its recitals, the Regulation promises to make things easier for companies, particularly small and medium-sized enterprises. It will no longer be possible for companies to be asked by multiple member states for a wide range of HTA data with a wide range of deadlines. The fact that this is explicitly mentioned in the recitals probably is due to the fact that the Regulation has been criticized in advance for a range of reasons, including the fear that, along with the MDR, it could lead to additional work for companies.
d) Have the promises been kept?
Whether the promises of reduced time and effort requirements and uniform assessments made by the HTA Regulation in its introduction have actually been kept is certainly open to question. Ultimately, a lot of the decisions are left to the member states themselves. Furthermore, the Regulation explicitly states that additional procedures may be conducted at the national level and that states can request additional data from developers for these procedures.
“If additional information, data, analyses and other evidence is needed for complementary clinical analyses, Member States should be able to ask the health technology developers to submit the necessary information, data, analyses and other evidence.”
HTA Regulation, Recital 15
This also raises the question of how much the HTA process as a whole will be delayed if the first step is taken at an EU-level and the second at the national level. In a 2018 statement, BVMed [in German] said it was expecting a 12-month delay.
So only time will tell whether the HTA Regulation will mean more or less work for companies.
e) Do manufacturers need to worry?
In brief: not at first. EU bodies will not start HTAs until January 2025. Priority in terms of the EU HTA will be given to medicinal products for the treatment of cancer. In addition, only a few HTAs will be conducted at first. It is therefore reasonable to assume that medical device and IVD developers will by and large be unaffected initially. However, medical device manufacturers should definitely keep an eye on developments in the next few years.
5. Summary and conclusion
The HTA Regulation attempts to standardize the HTA process at the EU level but makes a lot of compromises in the process. The Regulation makes clear in several places that the final decision on health technologies remains with the member states. This compromise had to be made because the EU treaties state that member states retain sovereignty over healthcare, but ultimately it weakens the effectiveness of the HTA Regulation. EU HTAs are limited to the clinical dimension and purely factual analyses. The assessment of the additional benefit and the consequences of the assessment (e.g., reimbursability) will continue to be national-level decisions.
However, this does not mean that EU HTAs are completely irrelevant.
Above all, the Coordination Group's reports will act as a basis for member states’ decisions. Member states will have to give due consideration to the EU reports when carrying out a national HTA (Art. 13(1)). This will be particularly noticeable when it comes to decisions on the use and reimbursability of a health technology in the German healthcare system according to SGB V.
However, only time will tell whether the HTA Regulation will ultimately mean more or less time and effort for health technology developers.

Author:
Dr. Anja Segschneider

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.