
Author:
Dipl.-Ing. (FH) Sarah Gruber
ISO 10993-17: What the standard has changed and now requires from manufacturers
The ISO 10993-17 standard is part of the ISO 10993 series of standards on biocompatibility. ISO 10993-17 describes the procedures for toxicological risk assessment.
In the fall of 2023, a comprehensive standard revision was published after over 20 years.
Medical device manufacturers should know,
- when they have to comply with ISO 10993-17,
- what the standard requires,
- what the current changes are, and
- when and how they should implement these changes.
This article will help.
1. Context of ISO 10993-17
1.1 The concept of toxicological risk assessment
Toxicological risk assessment (TRA) is an essential component of the biological evaluation. It is used to identify potential health risks and evaluate them by determining limit values and estimating exposure.
Health risks can arise from
- leachable substances (from the material itself),
- impurities (e.g., from the production of the medical device) or
- degradation products,
that enter the human body from the medical device.
The toxicological risk assessment is a prerequisite within the evaluation of biocompatibility according to ISO 10993-1 in order to ensure the legally required patient safety when using medical devices.
Further information
The article on biocompatibility and ISO 10993-1 gives you an easy introduction to the topic.
1.2 The regulatory requirements
The MDR formulates the legal requirements as follows:
Devices shall be designed, manufactured and packaged in such a way as to minimise the risk posed by contaminants and residues to patients, taking account of the intended purpose of the device, and to the persons involved in the transport, storage and use of the devices. Particular attention shall be paid to tissues exposed to those contaminants and residues and to the duration and frequency of exposure.
MDR, Annex I, Part 10.2
ISO 10993-1, for example, refers to the need for toxicological risk assessment:
Adequate risk assessment requires characterization of toxicological hazards and exposures, as well as other potential biological responses to medical devices.
ISO 10993-1, Section B.2.1
1.3 Terms and abbreviations
This article uses many concepts and definitions from ISO 10993-17. Here is an overview:
TCL | Tolerable Contact Level | Estimation of surface contact exposure to an identified constituent that does not cause significant irritation |
TI | Tolerable Intake | Estimation of daily exposure to an identified constituent over a period of time (e.g., acute, subacute, subchronic or chronic) based on the body mass considered to be of no health concern |
POD | Point of Departure | Low point on a toxicological dose-response curve established on the basis of experimental or observational data. Various values can be considered as POD, such as the low reference dose value, the lowest observed adverse effect, the minimum irritation or a value with no irritation or no observed adverse effect. |
EEDmax | Worst-case exposure dose per kilogram of body weight | The exposure dose describes the amount of a constituent that comes/can come into contact with the body via an exposure route during a certain period of time. |
MoS | Margin of Safety | Ratio between the tolerable contact level of the constituent (numerator) or the tolerable intake (numerator) and the exposure dose (denominator) |
TTC | Threshold of toxicological concern | Exposure level below which there is no significant risk to human health |
Tab. 1: Important terms and abbreviations from ISO 10993-17
1.4 Buy ISO 10993-17
You can find the English version of ISO 10993-17:2023 for around €30 on the evs.ee website.
2. Toxicological risk assessment procedure
2.1 Overview
Before the actual toxicological risk assessment (TRA), the manufacturer must identify the constituents of the medical device the patient may be exposed to during clinical use. In doing so, the manufacturer should proceed in accordance with the requirements of the sister standard ISO 10993-18.
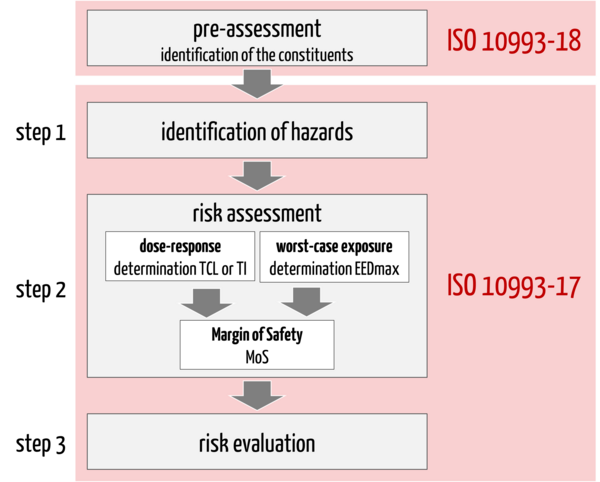
Only then does the actual TRA (scope of ISO 10993-17) take place, which can be divided into three steps (see Fig. 1):
1. Identification of hazards that may arise from the identified constituents of the medical device
2. Risk assessment including
- Determination of the tolerable contact level or intake (alternative: toxicological threshold of concern: TTC)
- Estimation of the exposure dose (worst-case approach)
- Derivation of the margin of safety (MoS)
3. Risk evaluation: The final step is the evaluation of the toxicological risk based on the identified hazards, the tolerable contact level or tolerable intake and the estimated exposure dose for each constituent. The risk estimates are compared with the acceptance criteria to derive possible gaps or residual risks.
In all steps of the TRA, manufacturers should consider the characteristics of the medical device to be evaluated and its clinical use:
- materials used
- type of use, e.g. cumulative use with new device
- contact type of exposure, e.g. skin
- exposure duration, e.g. chronic
- patient group, e.g. children
- dimension/contact area and number of medical devices in use
- uncertainty factors for the use of data from the literature
2.2 Pre-assessment: Identification of the constituents
In order to assess toxicological hazards, the manufacturer must identify the constituents that may be released during the clinical use of their medical device. This is done by means of a comprehensive material characterization of the medical device parts with direct and/or indirect patient contact.
Information on all constituents of the materials used is usually missing. A chemical analysis must then be carried out on the final medical device in accordance with ISO 10993-18. The clinical application is simulated by selecting a suitable extraction method, e.g. by taking into account the contact surfaces, contact type and contact duration.
Attention
The right choice of method in chemical characterization is essential for a successful toxicological risk assessment!
Example
100 % solvent, 72 h extraction at 50 °C is not an adequate simulation of a short-term application of a polymer to intact skin.
An incorrect choice of extraction medium, duration or temperature can lead to degradation of the material, which does not occur in the clinical application.
A large number of unidentifiable substances complicates the TRA and can lead to residual risks that may result in further investigations.
2.3 The actual TRA
Step 1: Identification of hazards
Once the manufacturer knows the constituents that may be released, the first step is to assess them:
- Do these constitutents need to be considered for the clinical use of the device?
- Do they pose any hazards?
The new version of the standard allows a Toxicological Screening Limit (TSL) to be used for this assessment. A TSL is used to evaluate the cumulative exposure dose of an constituent over a period of time and determine whether it poses a negligible toxicological risk. If the maximum exposure dose is below the specified TSL, no further risk assessment for systemic harm is necessary.
Example
If 100 µg of an identified constituent is extracted from a single medical device per day and a single medical device is in contact with the body for less than or equal to 30 days (no repeated use), the TSL is as shown in Table B.1 of the standard:
TSL = 120 x 1 = 120 µg
The cumulative exposure dose (for a single application of a device) corresponds to the detected 100 µg.
This means that the cumulative exposure dose is lower than the TSL. A further risk evaluation for systemic harm is not necessary.
Caution
The TSL approach is not intended for
- the cohort of substances of concern,
- insufficiently identified or irritating constituents,
- sensitive patient populations (young infants, i.e. six months or younger).
To identify the hazards posed by the substance released from the medical device, the first step is to collect and evaluate toxicological data on the substance (database research). Consideration of the clinical use and the quality of the data are important here:
- consideration of the identified endpoints based on categorization of the medical device according to ISO 10993-1
- consideration of the route of exposure, e.g. skin, oral intake, inhalation
- assessment of the quality of the data, e.g. by stating the guideline according to which the study was conducted
As an output of this step, the manufacturer knows the hazards of the identified constituent based on the available toxicological data from the literature.
Step 2: Risk assessment
The second step is the risk assessment. The manufacturer weighs up two things:
- the tolerable intake for the type of contact and duration
- the worst-case maximum exposure during use of the device
This requires several calculation steps.
a) Calculation of the tolerable contact level or tolerable intake
The tolerable contact level or intake of the identified constituent is determined. The tolerable contact level TCL or the tolerable intake TI is calculated. These describe the dose-response relationship of the constituent, i.e., the dose above which a toxicological effect is possible.
The collected toxicological data form the basis for the correct determination of TCL/TI. They provide the information up to which the exposure value of the contact or intake of the constitutent is assessed as safe.
This information is referred to as the Point of Departure (PoD). The following are possible:
- toxicological limit values of the constituent (e.g., the No Adverse Effect Level, NOAEL)
- data of a chemical surrogate (with justification and uncertainty factor)
- in silico data (e.g., use of TTC values of the Cramer Classes)
A conservative approach is recommended when determining TCL/TI. To this end, the manufacturer should take additional uncertainty factors into account (Modifying Factors, MF). These factors describe uncertainties arising from the transmission of data with different initial parameters:
Uncertainty factor | Explanation, example |
Intraspecies variation | Consideration of variations within the human population with regard to biological uptake, metabolism, tissue distribution, excretion and tolerance of substances |
Interspecies variation | Are data from studies with humans available or are they animal studies? Consideration of extrapolation of data from animal studies to humans |
Quality of the data | If the data from studies are carried out in accordance with the relevant guidelines and GLP (Good Laboratory Practice), it can be assumed that the data is of good quality. |
Quality of the study | Data are available for a different route or duration of exposure |
Exposure route and duration | If, for example, data from short-term studies are used for long-term exposure, this must be taken into account via safety factors. |
Limit values | The use of limit values such as Low Adverse Effect Level (LOAEL) instead of No Adverse Effect Level (NOAEL) must be taken into account. |
Tab. 2: Uncertainty factors that must be taken into account in toxicological data
Alternatively, manufacturers may choose a toxicological threshold of concern (TTC) for the substance if little or no toxicological data is available. The TTC is a value below which the risk of harm is considered negligible.
b) Estimation of the exposure dose
The next step is calculating each constituent's estimated worst-case exposure dose (EEDmax). Various factors are taken into account:
- bioavailability of the substance
- size and materials of construction of the medical device
- intended use
- route of exposure
- patient population
- duration and frequency of use
c) Derivation of the margin of safety (MoS)
A further step is required for the risk assessment: the calculation of the margin of safety (MoS) and, thus, the representation of the acceptability of the toxicological risk.
The MoS is determined by comparing the estimated exposure dose with the tolerable contact level (TCL) or tolerable intake (TI). An MoS value > 1 indicates a low toxicological risk.
Example
As part of a chemical analysis according to ISO 10993-18, a release of 150 µg/product/day of a constituent was detected. One device is used once per patient (short-term application < 24 h).
After consideration of toxicological data, it can be concluded that 100% of the constituent can be absorbed by the human body. The EEDmax results in a dose of 2.5 µg per kilogram of body weight for an adult patient with an assumed body weight of 60 kg.
EEDmax = maximum release (150 µg) / body weight (60 kg) = 2.5 µg/kg body weight
A limit value of 30 µg/kg bw/day was derived as the TI for the constituent using in silico methods.
This results in a MoS of 12:
MoS = TI / EEDmax = 30 / 2.5 = 12
With an MoS of 12, it can be assumed that there is a low risk to health from the release of the constituent.
Step 3: Risk evaluation
In the third step, the manufacturer decides on risk acceptance based on the criteria previously defined in accordance with ISO 14971 and ISO 10993-1. These criteria determine whether the estimated risk is acceptable or whether further measures are required.
Suppose the acceptance criteria are not met, for example, because the exposure to a constituent is too high. In that case, the manufacturer must further investigate the toxicological risk and determine additional risk reduction measures.
Example
Options for minimizing risk include, for example, changing the intended purpose, such as excluding certain patient groups.
Additional tests can be carried out as a follow-up activity, the output of which enables a more precise evaluation and the exclusion of risks.
3. Practical tips: What you should look out for in the TRA
Tip 1: Involve all data
Involve all data from the clinical use in the analysis. This includes:
- number and dimensions of devices per application
- patient group
- contact duration (cumulative, if applicable)
- type of contact
These points are important in order to be able to realistically assess the risks from released constituent when using the medical device and to address the endpoints in accordance with ISO 10993-1.
Tip 2: Sensible chemical characterization
Select a sensible analysis and parameters for chemical characterization, as this usually provides the main input for the toxicological assessment.
The derivation of the selected method should be based on the clinical use of the medical device and, at the same time, fulfill the requirements of ISO 10993-18. In this way, you avoid additional work by evaluating constituents that would not be released in the actual application.
Tip 3: Pay attention to the quality of toxicological data
Pay attention to the quality of the toxicological data used and document it. Animal studies are carried out in accordance with toxicological guidelines such as OECD and quality management systems such as GLP. Data from such studies can be classified as qualitatively appropriate and are suitable for the derivation of limit values.
Tip 4: Consider clinical use
When deriving limit values, always pay attention to your medical device's clinical application and consider this. In particular, you should know the exposure routes, as these have an influence on the limit values.
Tip 5: Estimate conservatively
Always use a conservative approach to ensure patient safety.
Examples
- If no data are available on the kinetic release of the substance during prolonged or repeated use, a continuous release of the initial amount per day must be assumed.
- Assume 100% for calculating the EEDmax if there is no clear data on the bioavailability of the substance in the literature.
- Always consider the largest possible number/contact area of the medical device during the intended use.
- When deriving the TI, always use the lowest POD and, if applicable, the more critical endpoint, e.g., chronic toxicity, for short-term use.
- Take into account a lower body mass if the device can also be used in patients with a lower weight (e.g., cancer patients).
Tip 6: Involve experts
Involve experts in planning the biocompatibility tests and have them carry out the toxicological risk assessment. Benefit from their experience when assessing the toxicological data. A lack of experience on the part of the assessors can lead to audit deviations, as this is a requirement of the standard.
4. The new ISO 10993-17:2023
a) The changes
An update to ISO 10993-17, which had been in force for over 20 years, was published in the fall of 2023. This means that the oldest standard in the ISO 10993 family, which was valid from 2002 to 2023, has undergone a significant revision.
The standard now no longer only deals with the definition of permissible limit values for substances but also describes how to carry out the toxicological assessment of a medical device constituent. The title of the standard has been adapted accordingly. The length of 65 pages can also be explained by the fact that the new version contains numerous examples.
Other new features include:
- comprehensive description of the process with calculation examples for determination of tolerable intake (TI), tolerable contact level (TCL), and worst-case exposure estimation based on body weight (EEDmax)
- implementation of approaches to reduce the assessment effort: TSL (Toxicological Screening Limit) and TQmax (total quantity released)
- selection of the initial value POD (Point of Departure) and use of uncertainty factors with specification of "default" values
- implementation of the Margin of Safety (MoS) as a calculation parameter specific to the endpoint
- explanations of the documentation effort (e.g., justifications for selection of the POD or uncertainty factors)
- removal of the Tolerable Exposure (TE) concept. Clinical application is now involved via exposure assessment.
b) The consequences
Good news: Toxicological risk assessment that were prepared for medical devices on the market before the revised standard came into force do not need to be adapted to the new requirements.
Nevertheless, check that your documentation is up to date and complete. Toxicological data on substances must be up to date. There may be changes to materials, suppliers, or the production process, new data, or relevant feedback from the market that need to be evaluated.
Please note
For new toxicological risk assessments, the update of the standard results in a significantly higher documentation effort. This can be kept manageable with a structured approach. Please refer to chapters 2 and 3 of this article and seek support if necessary (see below).
5. Summary and conclusion
The current edition of ISO 10993-17 introduces far-reaching changes. In particular, it describes a process for toxicological risk assessment and supports readers with many examples.
The toxicological risk assessment process consists of a total of three steps:
- identification of hazards
- risk assessment
- risk evaluation
Manufacturers who do not yet comply with the latest version of the standard should check that their documentation is up to date and complete (see chapter 4b).
Support
The Johner Institute team
- carries out the complete toxicological risk evaluation or supports manufacturers in doing so.
- helps with a gap analysis to ensure that the toxicological risk assessment is in line with the state of the art and that audits and approval procedures run smoothly.
- plans, organizes, and evaluates biocompatibility testing of medical devices, including chemical analyses for material characterization.
Contact us here if you would like support.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.