Performing FDA Recalls Correctly
If a medical device does not (or no longer) meets the FDA’s requirements, manufacturers, distributors and importers must recall it. This is particularly true if the device presents a hazard.
In this article you will learn:
- What a recall is
- What has to be considered when recalling a device
- What the difference between a “recall”, “correction” and “removal” is
- Who is responsible for the recall, correction or removal
1. What is a recall?
A recall is a way for manufacturers to correct their devices or remove them from the market if they do not (or no longer) comply with the FDA’s requirements. In this way, manufacturers and distributors fulfill their responsibility to protect public health and well-being against medical devices that present a risk of injury or gross deception or are otherwise defective.
Definition: Recall
“Recall” means a firm's removal or correction of a marketed product that the FDA considers to be in violation of the laws it administers and against which the agency would initiate legal action, e.g., seizure. Recall does not include a market withdrawal or a stock recovery.
Source: FDA website
What are the different types of FDA recall?
- Medical device recalls are generally initiated voluntarily by the manufacturer under 21 CFR 7.
- Recalls can also be ordered by the FDA. This happens when the manufacturer or importer does not voluntarily remove a device that is a risk to health from the market. The recall request is issued by the FDA exercising its medical device recall authority in accordance with 21 CFR 810.
- Corrections and removals according to 21 CFR 806 are closely related to recalls. They are also initiated by the manufacturer itself, for example, to ensure that the device continues to comply with the legal requirements. However, a correction or removal must be reported under certain circumstances.
2. Voluntary recall under 21 CFR 7 – Enforcement Policy
A medical device recall according to 21 CFR 7 is a voluntary action on the part of the manufacturer or distributor. A device available on the market must be recalled if it violates FDA regulations. This is particularly the case for devices that endanger public health and well-being because:
- they present a risk of injury,
- they involve gross deception or
- they are otherwise defective.
Additional information
See the FDA's guidance document on
Initiation of Voluntary Recalls Under 21 CFR Part 7, Subpart C
a) What recalls do NOT include
A recall under 21 CFR 7 does not include a market withdrawal or a stock recovery.
Definition: Market withdrawal
“Market withdrawal” means a firm's removal or correction of a distributed product which involves a minor violation that would not be subject to legal action by the FDA or which involves no violation, e.g., normal stock rotation practices, routine equipment adjustments and repairs, etc.
Source: FDA website
However, if the FDA deems that the withdrawn device does not comply with its regulations, the recall regulations apply. If a company is not sure whether a device breaches FDA regulations, it must report the market withdrawal to the FDA.
In addition, electronic products that emit radiation and are subject to 21 CFR 1003 and 1004 are not subject to the requirements of 21 CFR 7.
b) Requested recall under 21 CFR 7
If neither the manufacturer nor the distributor voluntarily recalls their non-compliant device and the FDA finds out, they can also request a recall under 21 CFR 7. Unlike a recall order under 21 CFR 810, a requested recall under 21 CFR 7 is intended for exceptional, urgent cases.
In this case, the request is sent to the manufacturer. The actions open to the FDA range from a simple request to seizure in extreme cases.
A request by the Food and Drug Administration that a firm recalls a product is reserved for urgent situations and is to be directed to the firm that has primary responsibility for the manufacture and marketing of the product that is to be recalled.
c) Voluntary recall process
Step 1: Notification of the recall to the FDA
A recall under 21 CFR 7 must be reported to the FDA. To do this, the manufacturer must contact its FDA Office of Regulatory Affairs (ORA) Division Recall Coordinator (DRC). Foreign manufacturers and importers must contact the DRC where their US agent is located.
The recall should be done as quickly as possible. No deadline is specified.
FDA health hazard evaluation
The FDA assigns a numerical class (I, II or III) to recalls to indicate the relative level of health hazard presented by the device being recalled.
- Class I - A situation in which there is a reasonable probability that the use of, or exposure to, a violative product will cause serious adverse health consequences or death.
- Class II - A situation in which use of, or exposure to, a violative product may cause temporary or medically reversible adverse health consequences or where the probability of serious adverse health consequences is remote.
- Class III - A situation in which use of, or exposure to, a violative product is not likely to cause adverse health consequences.
The FDA's internal process varies depending on the class assigned. All Class I recalls are posted on the FDA’s website. Therefore, the class should have an impact on the recall strategy.
For high-risk (class I) cases, manufacturers should establish stricter measures in their strategy, e.g., 100% recall effectiveness, 100% customer response rate, more attempts to contact all customers, notification in various media, etc.
Step 2: Establishing the recall strategy
The recalling firm should develop a recall strategy and this strategy must be approved by the FDA. It should be aligned with the potential consequences of the potential hazard caused by the device.
Definition: Recall strategy
"Recall strategy” means a planned course of action to be taken in conducting a specific recall, which addresses the depth of recall, need for public warnings, and extent of effectiveness checks for the recall.
Source: FDA website
The FDA lists the following factors that should be considered when designing the strategy:
- Results of the health hazard evaluation (the class influences the strategy. For example, companies must act more quickly and decisively in high-risk cases).
- Ease in identifying the device.
- Degree to which the device's deficiency is obvious to the consumer or user.
- Degree to which the device remains unused in the marketplace.
- Continued availability of essential products.
The recall strategy must take the following elements into account (depending on the severity of the breach):
- Depth of recall
The distribution chains to be covered by the recall have to be taken into account: consumer or user level or wholesale to retail level. The extent of the recall depends on the degree of hazard posed by the medical device. - Public warning – yes or no?
If there is a particularly serious hazard, a public warning may also be necessary. This has to be coordinated with the FDA, who will generally issue the warning itself. However, a public warning is reserved for urgent situations. - Effectiveness checks
The recalling firm must establish strategies for checking the effectiveness of the recall measures. According to the FDA, this can involve personal visits, telephone calls, letters, or a combination of these things.
Definition: Recalling firm
“Recalling firm” means the firm that initiates a recall or, in the case of a Food and Drug Administration-requested recall, the firm that has primary responsibility for the manufacture and marketing of the product to be recalled.
Source: FDA website
Step 3: Send the recall letter
A recalling firm must promptly notify each of its affected direct customers about the recall. The format, content, and extent of a recall notification should be in line with the hazard presented by the device. For their part, customers are obliged to comply with the recall request immediately.
The FDA gives some tips for writing the communication:
- Be brief and to the point.
- Clearly identify the product, size, lot number(s), code(s) or serial number(s) and any other relevant descriptive information to enable accurate and immediate identification of the product.
- Explain concisely the reason for the recall and the hazard involved, if any.
- Provide specific instructions on what should be done with respect to the recalled products.
- Provide a way for the recipient to report to the recalling firm whether it has the device.
- The recall communication should not contain irrelevant information, promotional materials, or any other statement that may detract from the message.
Step 4: Report on the recall status
The recalling firm must issue regular reports to the Division Recall Coordinator (DRC) on the status of the recall so that the FDA can assess the progress of the recall. In general, the reporting interval will be between two and four weeks.
d) Public notification and termination of a recall
Termination of a recall
A recall will be terminated when the FDA determines that all reasonable efforts have been made to remove or correct the device in accordance with the recall strategy. The DRC will send written notification of the end of the recall to the recalling firm. The firm can also request the termination of the recall itself.
Public notification of a recall
The FDA publishes all recalls in its weekly FDA Enforcement Report.
Additional information
- Regulatory Procedures Manual, Chapter 7 Recall Procedures
- Medical Device Class I Recalls
- Device Recalls: A Study of Quality Problems, document #273, contact: dice@cdrh.fda.gov
- “Evaluation of Software Related Recalls for Fiscal Years 1983-91”, May 1992, contact: dice@cdrh.fda.gov
3. Prohibition of use according to 21 CFR 810 – Medical Device Recall Authority
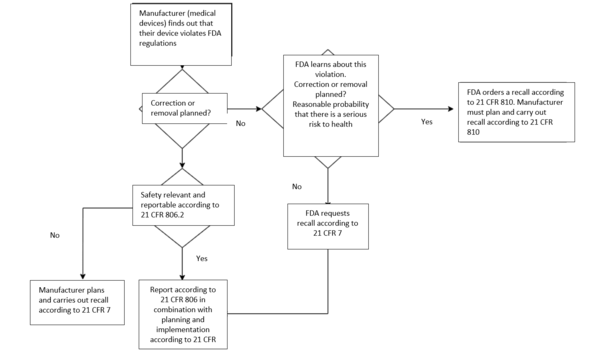
If medical devices do not (or no longer) meet the FDA requirements, they are generally recalled voluntarily under 21 CFR 7. In exceptional cases where the manufacturer or importer does not voluntarily recall a device that poses a health hazard, the FDA will take enforcement action under 21 CFR 810 – Medical Device Recall Authority.
This usually happens:
- after the FDA has given the company an opportunity to consult with it,
- if the company nevertheless does not carry out the recall voluntarily, and
- the FDA concludes that there is a reasonable probability that a device intended for human use would cause serious adverse health consequences or death.
In this case, the FDA first prohibits the distribution and use of the device and also notifies potential users, such as medical facilities. The prohibition of use can also later be changed to a “mandatory recall.”
If the conditions of 21 CFR 810 are met, it takes precedence over 21 CFR 7.
4. Corrections and removals according to 21 CFR 806 – Medical Device Corrections and Removals
a) Difference between recall, correction, and removal
According to 21 CFR 806, corrections are closely linked to recalls. The fundamental difference between a correction and a recall is that corrections may also be carried out even if the device does comply with the FDA regulations.
- Recall: A recall is a correction or removal that occurs when a device does not comply with the FDA regulations.
- Correction: The device is improved, adjusted, relabeled or repaired. This does not necessarily have to be a response to a breach of regulations.
- Removal: The physical removal of a device from its point of use to another location for repair, modification, adjustment, relabeling, destruction, or inspection.
So, a recall can be both a correction and a removal. However, it is always the result of a breach of FDA regulations. Corrections and removals can also be carried out regardless of whether there is a breach or not.
Definition: Correction
“Correction” means repair, modification, adjustment, relabeling, destruction, or inspection (including patient monitoring) of a product without its physical removal to some other location.
Source: 21 CFR 806.2 (d) and (j)) according to the FDA
Definition: Removal
“Removal” means the physical removal of a device from its point of use to some other location for repair, modification, adjustment, relabeling, destruction, or inspection.
Source: 21 CFR 806.2 (d) and (j)) according to the FDA
b) When a correction has to be reported
A correction does not normally have to be reported. However, it can become reportable if it is carried out in order to:
- Reduce a risk to health as defined by 21 CFR 806.2 (k) resulting from the use of the device. The definition of “risk to health” is the same as the definitions used for Class I and II recalls in 21 CFR 7.3(m) (see above)
- To remedy a breach of the FD&C Act caused by the device and which may present a risk to health, unless the information has already been provided in a reportable event notification (medical device report, MDR) in accordance with 21 CFR 803 (Medical Device Reporting) or the corrective or removal action is exempt from the reporting requirements (21 CFR 806.1. (b)).
- A report also has to be sent if the event is the result of a use error.
Manufacturers and importers must keep records of corrections and removals that do not have to be reported to the FDA. If a report is not required according to 21 CFR 806, the company can submit a voluntary report in accordance with 21 CFR 7.
Exemptions from the reporting requirement
The following actions are exempt from the above reporting requirements (21 CFR 806.1 (b)):
- Actions to improve the performance or quality of a device
- Market withdrawals that involve a minor violation of the FD&C Act that would not be subject to legal action by FDA or that involve no violation of the FD&C Act, e.g., normal stock rotation practices.
- Routine servicing (“any regularly scheduled maintenance of a device, including the replacement of parts at the end of their normal life expectancy, e.g., calibration, replacement of batteries, and responses to normal wear and tear”).
- Stock recovery
Safety warnings/field safety notices are not considered reportable corrections or removals, but may be remedial actions.
c) The correction and removal process
Who has to submit a report?
The company that initiates the correction must submit a report. This could be the manufacturer or the importer.
What deadlines apply?
The report must be submitted to the FDA within 10 working days from the date the company initiated the correction or removal.
This same deadline applies for changes to the original correction report (ten working days from the change).
What does the report have to include?
The information that the manufacturer or importer must include in the report is set out in § 806.10(c).
- Registration number, date of the report, a sequence number (001, 002, etc.), “C” for correction or “R” for removal
- Name, address, telephone number and contact for the company responsible for the correction or removal
- The brand name and common name of the device and its intended use
- The FDA marketing status, i.e., 510(k), PMA, preamendment status and device listing number
- Model/catalog number, lot/serial number
- Manufacturer's contact information (name, address, telephone number, contact person), if different from point 2 above
- A description of the event(s) and the corrective or removal actions that have been, or are expected, to be taken
- Any illness or injuries that have occurred with use of the device. If applicable, include the numbers of the medical device reports submitted under 21 CFR 803.
- The number of devices subject to the correction or removal
- The date of manufacture or distribution, and the expiration date or expected life
- Name, address and telephone number of all consignees (domestic and foreign) as well as the dates and number of devices distributed to each consignee
- A copy of all communications regarding the correction or removal
- A statement as to why any required information is not available and a date when it will be submitted
Who is the report addressed to?
Manufacturers and importers have two options for reporting corrections and removals:
- Via the FDA Electronic Submission Software (eSubmitter). This is the option recommended by the FDA.
- By email to the Division Recall Coordinator (DRC) of the FDA's Office of Regulatory Affairs (ORA).
5. Guidances and other important literature on FDA recalls
We recommend the following resources for further research:
- The FDA's web page on recalls and corrections/removals
- FDA guidance documentInitiation of Voluntary Recalls Under 21 CFR Part 7, Subpart C (PDF)
- Guidance for Industry and FDA Staff (PDF)
- On the FDA's CDRH Learn platform there are numerous presentations and webinars on the topics of recalls and corrections and removals in the Postmarket Activities / Medical Device Recalls section.
6. Tips
To ensure that the recall or correction process runs smoothly, we have put together the following tips for manufacturers and importers:
- Be aware of your responsibilities
Foreign manufacturers and importers should be prepared for recalls, corrections and removals. Companies are often under the mistaken belief that their US agent is responsible for the report. But recalls, corrections and removals are the responsibility of the manufacturer or importer. - Establish processes
Companies that sell their devices on the US market should establish a recall/reportable correction or removal procedure as part of their QM system.
The process flow should be practiced so that the company can react quickly in an emergency.
- Create report templates
Manufacturers and importers should create templates, e.g., for reports to the FDA, for reports to users, etc. This saves time in case of an emergency and helps to avoid errors. - Establish clear roles and responsibilities
Establish roles for recall, correction or removal procedures in advance. Establishing substitution rules is also important. It must not be the case that a reportable recall cannot be responded to because employees are on vacation. - Pay attention to 21 CFR 806
Manufacturers often only think about 21 CFR 803 (Medical Device Reporting) when it comes to reporting incidents. However, if manufacturers take field corrective actions in response to an incident, 21 CFR 806 (Medical Device Correction and Removals) must be followed. If companies fail to do so, they will receive a warning letter from the FDA.
- Learn about recalls and corrections/removals in Auditgarant
In the Johner Institute's Auditgarant, manufacturers and importers can find instructions and other important information on recalls and corrections/removals. Among other things, there is a template for recall and correction/removal procedures.
7. Conclusion
Manufacturers and importers in particular should make sure to familiarize themselves with the processes for recalls, corrections and removals. If there are any problems with their devices in the USA, they may be responsible for these procedures. Failure to act may result in a warning from the FDA and, in the worst case, a lawsuit.
At first glance, the processes for recalls, corrections and removals seem complicated and lengthy. But if you prepare well and follow the tips given above, everything should run smoothly, even in an emergency.

Author:
Luca Salvatore

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.