FDA Human Factors Guidances
The FDA describes its requirements for human factors engineering in two documents:
1. Applying Human Factors and Usability Engineering to Medical Devices (Feb 2016, final version)
2.Content of Human Factors Information in Medical Device Submissions (Dec 2022, draft)
This article
- summarizes the most important requirements for you,
- contains an annotated version of the second document for download, and
- provides assistance to help you meet these requirements quickly and safely and avoid problems during the approval process.
What the FDA calls "Human Factors Engineering" (HFE), IEC 62366-1 calls "Usability Engineering". The FDA defines the term as follows:
Definition Human Factors Engineering
Application of knowledge about human behavior, abilities, limitations, and other characteristics to the design of medical devices (including software), systems and tasks to achieve adequate usability.
Source: Content of Human Factors Information in Medical Device Submissions
1. How the FDA's "Human Factors Guidances" interact
In the introduction to the document "Content of Human Factors Information in Medical Device Submissions", the FDA describes its objectives:
- This document is intended to guide manufacturers on what human factors (usability) information must be submitted to the FDA for approval, depending on the risk of the product.
- It is intended to replace the FDA guidance "List of Highest Priority Devices for Human Factors Review".
- It is intended to supplement, clarify and even replace Chapters 3 (Definitions), 9 (Documentation), and Annex A (HFE/UE Report) of the FDA Guidance "Applying Human Factors and Usability Engineering to Medical Devices" (HFE Guidance).
- The HFE Guidance defines the HFE process that results in the documents that must be submitted in whole or in part to the FDA. The second mentioned guidance document determines which documents have to be submitted.
Both HF guidelines are relevant in (almost) all approval processes: 510(k), De Novo, PMA, and HDE.
Note
The FDA has published human factors engineering guidance documents for certain product classes. There is also a guidance document for combination products. Another article explains their requirements.
2. Human Factors Engineering: Which documents to submit
a) Determination of the category
The HF documentation depends on the risk of the device or the modification of an existing device:
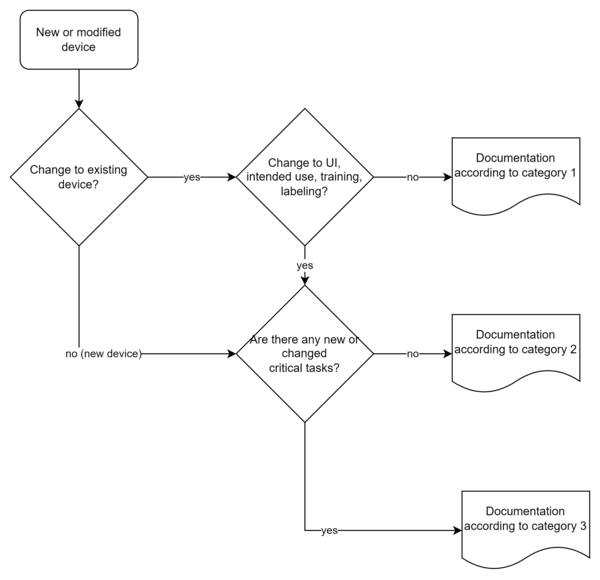
In Chapter VI (Examples), the FDA uses examples to describe how the respective case distinctions are to be made. Software in which the algorithms are changed but not the user interface is classified as a change for which category I documentation is sufficient.
b) Determination of the documents to be submitted
The categories determine the scope of the HF documentation to be submitted:
| Documents | Cat. 1 | Cat. 2 | Cat 3. |
| Summary | X | X | X |
| Description of intended use (incl. user, context of use, training) | X | X | |
| Description of the user interface | X | X | |
| Summary of known usage problems | X | X | |
| Summary of the preliminary evaluations | X | ||
| Risk analysis (focus on usability) (incl. hazards, risks and description of critical tasks) | X | ||
| Usability validation details | X |
In the section "Recommended content of human factors information in marketing submissions", the FDA describes in more detail the documents listed in the table.
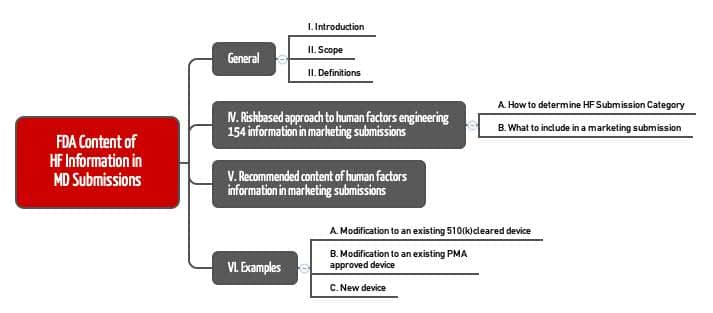
Note
The "Critical Tasks" largely correspond to the "Hazard Relevant Use Scenarios" of IEC 62366-1. The FDA now defines "tasks" as "One or more user interactions with a medical device to achieve a desired result".
The FDA would like to see a clear statement on the presence or absence of critical tasks, as well as a description and assessment of the residual risk. This can speed up the FDA's approval process, as this information does not have to be requested subsequently.
Download
An annotated version of the FDA document on documents to be submitted can be downloaded here.
3. Human Factors Engineering: The HFE process
a) The guidance documents
The FDA's document "Applying Human Factors and Usability Engineering to Medical Devices" comprises 49 pages, ten chapters, and four appendices.
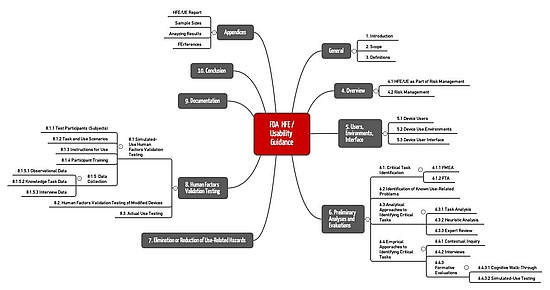
This document contains definitions and an introduction to the subject in Chapters I to IV. The FDA describes concepts such as risk management, user, use environment, and user interface.
It also describes the activities that manufacturers should go through as part of usability engineering:
- Risk analysis and identification of critical tasks (e.g., with FMEA, task analysis, analysis of known usage problems, heuristic evaluation, expert review)
- Formative evaluation (partly with comparable methods)
- Elimination of usage-related hazards
- HF Validation Testing
Note
The requirements of IEC 62366-1 are very similar to the requirements in this guidance document, even if the terminology does not quite match. For example, the standard does not speak of Human Factors Validation Testing, but of Summative Evaluation.
In contrast to IEC 62366-1, the FDA hardly addresses the process. On the other hand, it describes the activities and methods in detail in the HFE Guidance. The latter are presented in a similar form in TR IEC 62366-2.
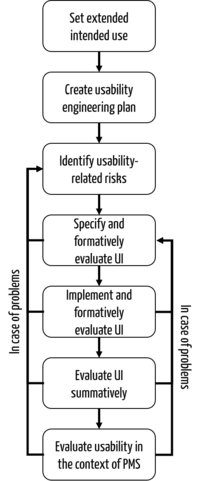
b) Implementation
A usability engineering process that meets both FDA and IEC 62366-1 requirements could be designed as follows:
Further information
The Johner Institute supports medical device manufacturers in FDA-compliant usability engineering and helps to achieve approvals quickly and safely in all parts of the world. To this end, our experts conduct usability studies in Germany, the USA, and other countries.
Read more about how our usability services contribute to your products' fast and safe approval.
4. Summary and conclusion
The FDA's guidance documents on usability engineering are easy to understand and follow. The risk-based approach to compiling the documentation to be submitted helps to speed up approval procedures without endangering the safety of patients due to a lack of usability.
Attention
As with the FDA's software documentation requirements, the guidance document "Content of Human Factors Information in Medical Device Submissions" determines the amount of documentation to be submitted, not the amount to be produced.
It is very useful to medical device manufacturers that IEC 62366-1 and the FDA's HFE guidance are now highly aligned. This allows, for example, formative and summative evaluations to be conducted in a manner that meets the requirements of both legal areas.
The Johner Institute is happy to help ensure that the resulting documentation meets the regulatory requirements.
Change history
- 2023-09-15: Reference to the FDA guidance document on combination products added to chapter 1

Author:
Dr. Nils Becker

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.