The EU Regulates Medical Laboratories – Are Laboratory Developed Tests Still Allowed?
This article will help you ensure that you will still be able to offer in-house IVD (also called laboratory developed tests, LDTs) under the IVDR and will explain the three options open to you avoiding legal disputes.
In-house IVD are a type of in vitro diagnostic test. But do regulatory requirements such as the IVDR also apply to these kind of products? This article provides an overview not just for medical laboratories.Fig.1: Laboratory Developed Tests: EU

1. Why you might have to reduce the range of tests you offer
The EU legislator is regulating laboratory developed tests for the first time with EU Regulation 2017/746 on In Vitro Diagnostic Medical Devices(IVDR). As a result, its requirements apply directly to laboratories that offer tests they developed themselves.
Most of these requirements are not new, but the additional requirements mean that you, as a medical laboratory, are no longer allowed to offer certain LDTs. For example, this can be the case if a manufacturer offers a comparable CE-IVD device on the EU market.
What this means for you and how to avoid this problem will be explained later in this article. But first let us address the basics.
2. In-house IVD: What are they?
a) Definition of terms
The IVDR does not define in-house IVD or LDT under the definitions in Article 2 but provides the following description in Article 5:
Description of in-house IVD in the IVDR
“devices manufactured and used only within health institutions established in the Union”
Source: IVDR
The guidance document MDCG 2023-1 of the Medical Device Coordination Group designates corresponding tests as "in-house devices":
Definition: In-house device
„A device that is manufactured and used only within a health institution established in the Union and that meets all conditions set in Article 5(5) of the MDR or IVDR.“
Source: MDCG 2023-01
The US FDA defines the term "Laboratory Developed Tests" as follows:
Definition: Laboratory developed test
“A laboratory developed test (LDT) is a type of in vitro diagnostic test that is designed, manufactured and used within a single laboratory.”
Source: FDA
b) Differentiation from medical devices manufactured in-house
LDTs, or ‘in vitro diagnostic medical devices from in-house production’, must themselves be differentiated from ‘medical devices from in-house production’. In the case of a ‘medical device from in-house production’, we would talk about in-house production according to Article 5(5) MDR.
Additional information
Read more here on the in-house production of medical devices, the definition of the term own production, and the difference between placing on the market, parameterization and combination.
c)Where in-house IVD are used
Medical laboratories examine specimens derived from the human body: Tissue and body fluids (e.g., blood, urine, cerebrospinal fluid, etc.). To make diagnoses or to obtain information relevant for the diagnosis, laboratories use:
- Commercial in vitro diagnostic (IVD) medical devices, or
- non-CE-marked, modified, or self-developed tests, known as LDTs
Anywhere CE-IVD devices are used, LDTs can be used as an alternative. However, the IVDR limits the use of in-house IVDs to what it calls health institutions and defines such institutions as follows:
Definition: Health institution
“‘health institution’ means an organisation the primary purpose of which is the care or treatment of patients or the promotion of public health;”
Source: IVDR
Medical laboratories provide patients or the physicians treating them with medical information for a diagnosis. That is why medical laboratories can be considered health institutions according to the definition.
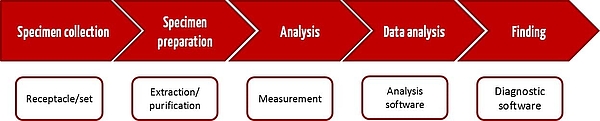
Each IVD test in a medical laboratory begins with a specimen being taken from the patient. Ideally, it ends with a result that is helpful for deciding on further treatment. Figure 1 shows the usual steps in an IVD test process from the taking of the specimen through to the results. CE-IVD devices or in-house IVD devices can be used for all these steps.
d) Criteria that make a test an LDT
An LDT is not usually a self-developed product but rather a procedure that uses self-developed devices in the test sub-steps. However, it is not usually clear to the patients or the physicians treating them whether a test is carried out with an LDT or a CE-IVD device (see Figure 2).
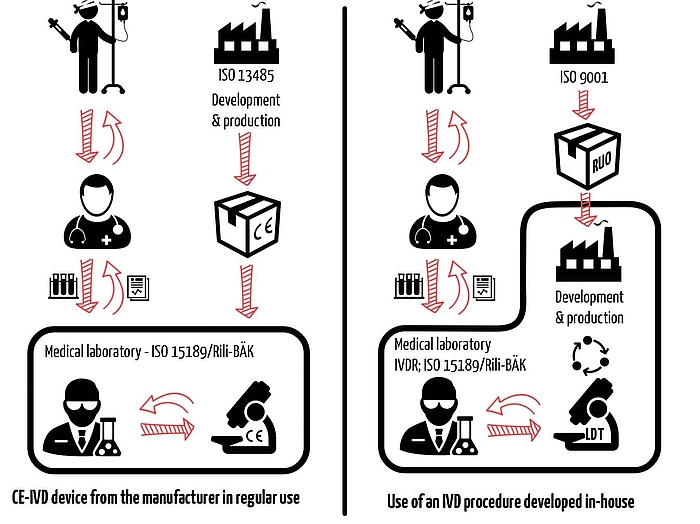
For this reason, a medical laboratory creates an LDT when it:
- uses products or procedures it has developed itself,
- uses non-IVD devices in diagnostic procedures (e.g. for research use only, RUO),
- combines products or processes that are not intended for the combination, or
- uses a CE-IVD device outside its intended purpose, e.g., when they:
- misuse the device (off-label use) or
- modify commercial devices (including if they deviate from the instructions for use of conventional IVD devices).
If one or more of these criteria apply to your tests, the requirements in the next section apply in full to your medical laboratory.
3. Legal requirements
a) Previous requirements for in-house IVD in Germany
Putting in-house IVD into service was only permitted according to section 12 of the MPG if the essential requirements of EU Directive 98/79/EC on In Vitro Diagnostic Medical Devices (IVDD) have been met. This means that if you have already offered an LDT before May 26, 2022, you must comply with Annex I of the IVDD.
Medical laboratories also have to comply with the “Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen” the Rili-BÄK for short. This is established in the Medizinprodukte-Betreiberverordnung (MPBetreibV). This has not changed since the IVDR came into force.
Careful!
Some laboratories with LDTs assume that the requirements have exploded with the IVDR. A closer look shows this is not true, because the requirements for in-house IVDs and their operators have been around for many years, including in the MPG as well as the MPSV, MPV, MPBetreibV and, indirectly, through the reference to the essential requirements, in the IVDD as well.
b) Requirements of the IVDR for in-house IVDs:
The IVDR wants to continue to permit in-house devices without the involvement of notified bodies and without CE marking (clauses 28 and 29 in the foreword section). As a result, in Article 5(5), the IVDR sets out the requirements that laboratories must meet for tests manufactured and used only within health institutions (this also includes the remote use of in-house IVD software; see MDCG 2023-1 3.2.2). These include:
- For all in-house IVDs, conformity with the essential safety and performance requirements according to Annex I of the IVDR must be demonstrated.
- The manufacture and use of the devices occurs under appropriate quality management systems.
- Neither Rili-BÄK nor ISO 15189 establish requirements for the development and manufacture of IVD devices. They also only provide incomplete specifications for the validation of these devices. As a result, it can be concluded that the requirements of section 7 of ISO 13485 apply.
- The health institution justifies in its documentation that the target patient group's specific needs cannot be met, or cannot be met at the appropriate level of performance by an equivalent device available on the market.
- This is possibly the most significant new requirement because LDTs for which there is an equivalent CE-IVD device on the market no longer have a right to exist.
Further requirements from Article 5 (5), such as
- a publicly available declaration,
- the surveillance of the devices,
- the prohibition of distribution and
- the prohibition of industrial-scale production
were already required by the MPG and the associated ordinances.
The Johner Institut supports medical laboratories in the implementation of all IVDR requirements. Contact us if you have any questions.
Further information
ISO 15189 was fundamentally revised at the end of 2022. It sets out more detailed requirements and emphasizes the importance of risk management even more strongly. It also incorporates content from ISO 22870 on point-of-care testing (POCT), which has been withdrawn.
c) FDA requirements for laboratory developed tests
At this point, it is worth taking a look across the pond because the FDA has been considering the regulation of LDTs since at least 2010, but has not yet been able to fully enforce anything. It is aware of the risks posed by incorrect or inappropriate treatment of patients that can result from the use of insufficiently controlled “high-risk LDTs”. The FDA no longer considers the previous “enforcement discretion” to be adequate for all LDTs.
Therefore, in 2014 the FDA published a draft “FDA Notification and Medical Device Reporting for Laboratory Developed Tests (LDT)” and collected feedback. This flowed into a “Discussion Paper” which the FDA published in January 2017.
In this document, the FDA wrote that it considers it problematic that a test developed by an IVD manufacturer is regulated differently to an identical test developed by a laboratory. This would undermine its risk-based approach. Surveillance should be risk-based and not dependent on who develops the test.
Exemptions
The risk-based approach and feedback that the FDA received to its “notification” led to some LDTs being exempted from the FDA's surveillance, including for example:
- Low risk LDTs;
- LDTs for rare diseases;
- LDTs intended solely for forensic use.
And other exemptions.
d) Summary of the legal requirements
Almost all requirements found in the IVDR already existed in the same or a similar way within the framework of the MPG and the associated ordinances.
Although the new Annex I of the IVDR is longer than Annex I of the IVDD, the key elements still include:
- Risk management
- A safe and powerful device design
- Usability,
- Performance evaluation
- software life cycle processes
IT security is a major new requirement.
Careful!
Completely new and therefore the “biggest chunk”: If there is an equivalent CE-marked IVD device on the market that has the same performance level as the in-house developed device, the CE-IVD device must be used (Article 5(5) d). In such a case, there is no longer a legal basis for the use of the device developed in-house and so the LDT can no longer be offered.
Exemptions from surveillance for certain LDTs (e.g. LDTs for the diagnosis of rare diseases), as implemented by the FDA, are currently not planned.
4. Transitional periods
In January 2022, new transitional periods were included in the IVDR, which also addresses the requirements of Article 5 (5) and thus affect in-house IVDs.
Important
The requirement for compliance with Annex I (Article 5 (5) sentence 1) has been in place since May 26, 2022, and has not been postponed.
5. Surveillance by authorities
In Germany, the respective State authorities are responsible for the surveillance of the laboratories for compliance with the IVDR. They also check whether laboratories are complying with the MPBetreibV and thus explicitly ensure compliance with the Rili-BÄK. If your laboratory is accredited according to ISO 15189, DAkkS is responsible for monitoring the implementation of the standard.
The rights of member states in the monitoring of health institutions are explicitly mentioned in IVDR Article 5 (5) subparagraph 2.
MDCG 2023-1 mentions in Chapter 3.7 information that can be consulted by the monitoring authorities.
6. Conclusion
a) The IVDR makes concrete demands on laboratories
Although the MPG already required the essential requirements of Annex I of the IVDD to be met, comprehensive compliance with these requirements was not achieved.
The IVDR now establishes uniform requirements for laboratories across Europe.
- Laboratories must develop, manufacture and surveil their own laboratory developed tests (largely) in conformity with the requirements of the IVDR.
- The use of commercially available IVD devices is preferred and laboratories can only offer their own LDTs if the devices on the market do not meet the required performance.
The previous system was also difficult to understand: why should the regulatory requirements for a laboratory test depend on whether it was developed by a laboratory or an IVD manufacturer?
A lot of medical laboratories are not fully aware of the existing and the new requirements. It’s high time they got busy. Tougher competition between laboratories and legal disputes with IVD manufacturers should be expected.
b) Your next steps as a medical laboratory
- Prioritize the fulfillment of the requirements from Annex I and implement them NOW, if you have not already done so.
- Expand your quality management system to include the required points and generate the resulting documentation by May 2024.
- Comply with the additional requirements of Article 5 (5) of the IVDR.
- Investigate the market and look for devices whose performance promises for the same patient target group corresponds to your in-house IVD.
There is no such device on the market? Congratulations, then you can offer your in-house IVD beyond 2028!
However, if there is an equivalent device on the market with the same level of performance, you should consider a strategy. The Johner Institute will be happy to support you.
Make a decision:
- No longer offer the test
- Purchase and use the authorized competitor test or
- Place your own test on the market as a CE-IVD device
If you decide to place the test on the market as a CE-IVD device:
- Establish a QMS according to ISO 13485
- Find a notified body
- Comply with the extended requirements of the IVDR(Depending on the risk class of the device) create device-specific technical documentation and declare conformity.
Tip
Get your free copy of the Johner Institut’s checklist for Annex I of the IVDR in our starter kit.
Note
The IVD team will support you as a medical laboratory in your adjustment to the IVDR and help you ensure that you comply with the requirements for LDTs. For example, we can hold an individual workshop to create the conditions needed to make your company and your devices fit for the future or review your documentation.
In the seminar “IVDR for medical laboratories”, our laboratory experts will give you a comprehensive overview of the requirements of the IVDR for the use of in-house IVDs. You will learn how to operate your existing in-house tests in a compliant manner and, after completing the seminar, you will be able to efficiently document new in-house IVDs in compliance with the IVDR using a developed roadmap.
Change history
- 2023-01-12:
- Adaptation of the definition "In-house device" according to the new MDCG 2023-1.
- Reference to MDCG 2023-1 and slight adjustments in section 3. b)
- Reference to MDCG 2023-1 in section 5.
- 2022-12-09: Notes on the revision of ISO 15189 added
- 2022-06-08:
- Conversions due to the IVDR which has become valid.
- "LDT" replaced by "In-house IVD", the official wording of the EU
- Adjustments due to the transition periods
- Removal of section 3 e) "Effects of the ongoing coronavirus pandemic"
- 2021-04-22: Section 3. e) on the effects of the ongoing coronavirus pandemic inserted. Editorial changes.
- 2021-12-01: Corrections, adjustments to links and editorial changes throughout the article. Adaptation of the sections
- a): Revision of the definitions
- b): Restructured
- 4: New section on transition periods added
- b): Adaptation to possible new transition periods

Author:
Ulrich Hafen

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.