Importers: What Do the MDR and IVDR Actually Demand?
The EU regulations the MDR and IVDR set out precise requirements for importers. And they also define who is an importer.
Bringing a device into the EU does not always constitute an import. And, furthermore, companies that import medical devices don’t just have to meet the requirements established for importers.
Tip
You will find below a link to a free checklist for importers that will help you quickly check and ensure conformity.
1. What is an importer?
a) Definition of the term “importer”
The MDR and IVDR define an importer as follows:
Definition: Importer
"any natural or legal person established within the Union that places a device from a third country on the Union market”
MDR Article 2, IVDR Article 2
b) Definition of the term “placing on the market”
The EU explains what it means by “placing on the market” in its Blue Guide:
A product is placed on the market when it is made available for the first time on the Union market.
Blue Guide
What is meant is making a specific device available that can be identified, for example, by a UDI-PI, such as an ECG with a specific serial number. It does not mean making available a type of product that could be identified by the UDI DI, for example, the ECG "Cardiomaster NT."
The definition in the Blue Guide matches well with the definition from the MDR and IVDR:
Definition: Placing on the market
“the first making available of a device, other than an investigational device, on the Union market”
MDR Article 2
Additional information
Note the important additional comments in the article on placing on the market.
c) Difference between an importer and a distributor
The Blue Guide makes clear that only manufacturers and importers can place devices on the market.
The operation [comment: it is referring to the placing on the market] is reserved either for a manufacturer or an importer, i.e. the manufacturer and the importer are the only economic operators who place products on the market. When a manufacturer or an importer supplies a product to a distributor or an end-user for the first time, the operation is always labeled in legal terms as ‘placing on the market’. Any subsequent operation, for instance, from a distributor to distributor or from a distributor to an end-user is defined as making available.
Blue Guide
An importer generally buys a medical device from a manufacturer (outside the EU) and sells it either to a distributor or an end user. In the case of direct sales, distributors who purchase medical devices outside the EU become importers.
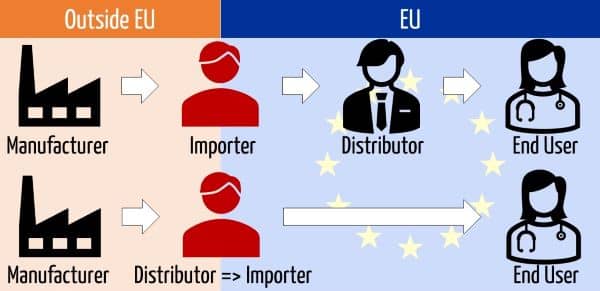
The MDR differentiates between distributors and importers. The book “Die Medizinprodukteverordnung (EU)” describes importers as a special type of distributor.
d) Difference between a manufacturer and an importer
Importers that modify devices or place them on the market under their own name are under the same obligations as manufacturers. Article 16 of the MDR specifies the cases in which these additional obligations apply.
Additional information
The article on distributors specifies and explains the additional obligations for these economic operators.
e) Essential and non-essential factors
It is essential that:
- The importer is based within the EU
- A transfer or rights takes place (between the importer and another natural or legal person) based on an offer or an agreement
In contrast, it does not matter whether:
- The agreement is made in writing or orally
- A payment is made for the transfer of ownership or it is done free of charge
- A transfer of ownership takes place. Also possible is the transfer of possession or any other rights (except intellectual property rights) ,e.g., by way of loan
f) What is not classed as an import?
The Blue Guide gives some examples of what is not classed as an import:
- A consumer buys a device outside the EU and brings it into the EU for their “personal use”
- A manufacturer based in the EU produces a device outside the EU but under its own QM system and then brings this device into the EU
- A device is brought into the EU for testing (verification, validation)
- A device is brought into the EU for a trade fair or exhibition. This requires the device to be labeled accordingly
2. What do the MDR and IVDR require from importers?
a) Registration in EUDAMED
Before importers import devices, they must register in EUDAMED. This requirement is established in Article 30 and Article 31 as well as Annex VI of the MDR. The requirements of the IVDR are identical.
Entries are publicly accessible and must be updated as required and confirmed every two years.
b) Their name on the device labeling
Article 13 of the MDR requires importers to “indicate on the device or on its packaging or in a document accompanying the device their name, registered trade name or registered trade mark, their registered place of business and the address at which they can be contacted, so that their location can be established”.
c) An inspection of the devices
The MDR and the IVDR require importers to inspect the devices to ensure:
- The device has been CE marked
- A declaration of conformity has been drawn up
- An authorized representative has been named
- The device is labeled in accordance with the MDR/IVDR
- Instructions for use (as required) are provided with the device
- The device has been assigned a UDI
However, there is no general testing obligation. So, importers do not have to test the medical devices.
It is currently being disputed whether the importer must carry out a complete inspection of the above aspects. Unlike for distributors, the MDR does not establish any exemptions (e.g., sampling). However, based on the Blue Guide, we assume that a complete inspection is not what is intended.
d) To contribute to the communication between manufacturers, users and authorities
Importers are an essential node in the communication between manufacturers, users and authorities. They must help ensure that all information required to ensure patient safety and that action can be taken quickly in the event of risks is available to these groups:
- Inform the manufacturer or its authorized representative if it considers or has reason to believe that the device does not comply with the MDR (Article 13(1) and Article 13(7)).
- Inform the authorities in the event of serious risks (Article 13(2))
- Provide the manufacturer, authorized representative and distributors with customer complaints (Article 13(6) and Article 13(8))
e) Transition periods
The MDCG clarifies in MDCG 2021-25 that economic operators must also comply with their obligations under MDR for legacy devices (Article 120, valid MDD certificate). For each economic operator, the MDCG lists the relevant articles of the MDR that they must comply with. For importers, these are the requirements of Article 13:
- Article 13(2) last paragraph: This deals with the information requirements in the context of post-market surveillance.
- Article 13(4): Importers shall verify that the product is registered in EUDAMED.
- Article 13(6): Importers shall keep a register of complaints.
- Article 13(7): This also deals with the information obligations and cooperation in corrective actions.
- Article 13(8): This also deals with information obligations and cooperation in corrective measures.
- Article 13(10): This paragraph concerns the cooperation of manufacturers with the authorities.
3. What importers should do now
a) Use our checklist
Importers should go through the Johner Institute's checklist for importers. It makes it quick and easy to check conformity with the MDR and/or IVDR requirements and to correct any potential deviations. This provides legal certainty and will ensure you avoid sanctions from authorities and courts.

b) Act now
NB!
There is no transitional period for importers of legacy devices. Since May 26, 2021, they have had to meet the requirements of the MDR. The Irish HPRA has made this clear in a notice.
c) Check whether they have to comply with additional requirements
If importers make modifications to the devices, they then have to comply with the requirements of Article 16. This applies, for example, if they:
- place devices on the market under their own name,
- change the intended purpose or
- translate the instructions for use.
In these circumstances, the UDI requirements, which guideline MDCG 2018-6 describes in more detail, also apply to importers.
4. Conclusion
The MDR and IVDR have significantly increased the requirements for importers. The EU has done this to try to ensure the traceability of devices and reduce the risks to patients posed by counterfeit and non-compliant devices.
For importers, this means additional time and costs, particularly when it comes to ensuring compliant device inspection with a quality assurance system and communication with authorities, manufacturers, distributors and third parties.
The Johner Institute’s checklist will help you quickly determine your own conformity, and detect and eliminate deviations.
Change history
- 2021-10-25: Chapter 2. e) inserted with the transition periods according to MDCG 2021-25.
- 2023-07-10: Revised Chapter 1. e) clarifying transfer of ownership as a requirement.

Author:
Luca Salvatore

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.