Regulatory Science: Europe is Flying Blind
In German-speaking areas, regulatory science is rarely practiced systematically.
And this failure creates problems for medical device manufacturers and harms both the health system and the region as a business location. It's high time the situation was changed!
1. What is regulatory science?
a) Definition
“Regulatory science” is a science that develops the (technical) principles, processes, methods and tools for:
- Formulating regulatory requirements that ensure the safety, performance and effectiveness of medical devices based on these principles, processes, methods and tools
- Understanding and anticipating the economic and other implications of these regulatory requirements on the healthcare system and the economy
A short and slightly simplistic definition could be:
Definition: Regulatory science
“Regulatory science consists of the application of science to support regulatory activity, particular the setting of regulatory objectives.”
Based on source
b) Difference between regulatory science and regulatory affairs
In contrast to regulatory affairs, regulatory science does not deal with:
- Making sure the wording and publication of regulations (laws, ordinances, guidelines) is legally watertight
- How these regulations are monitored (e.g., audited) and enforced
- Which documents manufacturers have to provide and in what form
Conversely, scientifically evaluating the logic and effectiveness of regulations is not part of regulatory affairs.
Additional information
Read more on regulatory affairs here.
2. Why we need regulatory science
a) Otherwise regulatory bodies are flying blind...
The bodies establishing regulations should know what the consequences of their actions are. But a lot of regulators (legislators, regulatory authorities) fail to meet this basic demand. They can’t know either since they don’t have the scientific basis.
Asking a few experts is not enough to build up this basis. A manufacturer who used a similar method, i.e., based primarily on interviewing patients or experts, would be reproached by the authorities and notified bodies for a lack of evidence. And rightly so. Eminence does not replace evidence.
Every law, every ordinance, every guideline has side effects. Designing these regulations in a way that optimizes the benefit-risk ratio requires an understanding of the economic and technological interdependencies. Researching these interdependencies is a key task of regulatory science.
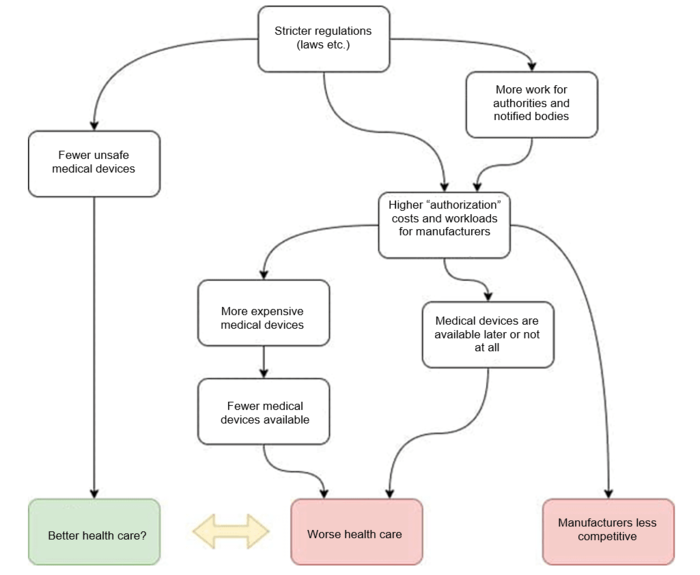
Conclusion: If they do not have an understanding of these interrelationships, legislators do not have an adequate basis for their decision-making. They are regulating completely blindfolded.
b) … And the regulations harm markets and patients
Regulations are generally introduced with the best of intentions. It is true that laws and other regulations in some countries are also used to close off their markets. Nevertheless, (hopefully) their primary goal is to contribute to optimal healthcare through safe and clinically effective devices.
But the legislators do not know the answers to key questions:
- Will, for example, the MDR and IVDR achieve this goal better than the previous EU directives (MDD, IVDD and AIMDD)?
- How many deaths and seriously injured patients does the EU Commission hope to prevent through the new regulations?
- And how many deaths and seriously injured patients caused by devices that the MDR and IVDR have prevented does the EU think there are?
What database did Brussels base its decisions on? Decisions that directly affect the European MedTech market worth over €120 bn per year and indirectly affect the healthcare market as a whole, which generates €1 bn in added value in Germany – every day! [Source]
The FDA goes so far as to claim:
In the U.S., FDA-regulated products account for about 25 cents of every dollar spent by American consumers each year — products that touch the lives of every American every day.
c) Otherwise we be dependent on, e.g., the FDA
The FDA is actively involved in the field of regulatory science. The “Advancing Regulatory Science” website provides an overview of its activities.
The easiest European approach might be (and probably is) to use these results for its own benefit without having to engage with the issue itself and without investing in regulatory science itself. But there are arguments against taking this easy option:
- Unnecessary dependence
Relying on these results means being dependent. The FDA could refuse access to this data at any time. - Limited transferability of data
The FDA's focus is justifiably on the USA. Its concern is the safety of US patients. And not all the results can be transferred to Europe. We don’t have identical device classes in Europe, nor do we have a population that is comparable in all aspects. The contexts of use (e.g., user, working environments) cannot be unthinkingly transferred to Europe. - Suboptimal evidence
Science does not stop at national borders. On the contrary, science, like regulatory science, is more successful when it has access to numerous resources, in particular a lot of data and scientists, worldwide. So, it is not about Europe versus the USA or versus the rest of the world. The aim is to create the broadest possible database and to gain as much knowledge as possible.
d) Otherwise manufacturers miss opportunities and lose time and money
The job of regulatory science isn’t just to prevent unsafe devices. It is also to promoting innovation and giving manufacturers new options for:
- Demonstrating the effectiveness and safety of their devices to authorities and notified bodies more easily, credibly and quickly (e.g., using computer models)
- Reliably detecting risks posed by their devices during development as well as once they have been placed on the market before they cause any harm to patients
- Being able to track the state of the art
- Finding out about new and alternative processes, methods, materials and tools to quickly develop better devices and gain a market advantage
3. Regulatory science should answer these questions
In order to achieve the above goals, regulatory science should provide answers to all relevant questions from all stakeholders.
a) Questions that regulators (should) ask themselves
Questions on the effects on the medical device market (incl. manufacturers)
- What will it cost manufacturers to implement a new regulation?
- How much damage to health will a new or amended regulatory requirement help to prevent?
- How manufacturers will be able to meet this requirement? And how long will it take them to do so?
- What percentage of manufacturers will fail to overcome this hurdle? Which medical products will not be available as a result or have their availability delayed?
- What are the consequences of these delays or of the device not being placed on the market for the health system?
- In what way do regulations harm small companies and/or innovation? How can regulations actually promote innovation?
- What disadvantages can new regulations lead to for a location? How can the location be strengthened by regulations?
Questions on the effects on authorities and notified bodies
- How long will it take for authorities and notified bodies to be able to monitor and enforce these requirements?
- What bottlenecks could they lead to for these stakeholders? (personnel, training, availability)
- Are they able to effectively enforce the requirements with manufacturers outside the EU as well?
- Which tools can/should authorities and notified bodies use to monitor compliance with the requirements as efficiently and effectively as possible?
- Which authorization procedures are available for which devices?
Questions on the effects on patients
- To what extent can regulations act in the interest of patients? Where do they interfere inappropriately with an individual patient’s freedom of choice and risk acceptance?
Questions on future legislation
- What trends do we have to be prepared for? Which devices, technologies, methods and materials will new regulations be needed for in the future?
- Which current regulatory requirements are no longer state of the art?
- Where are regulations that would be necessary to improve healthcare, e.g., through safer devices, missing? Where do regulations hinder and damage the location or even the health system?
- How can the regulatory framework be designed so that, on the one hand, the state of the art is adopted quickly enough and, on the other, the legislation can keep up with the speed of change?
- How is it possible, on the one hand, to word the regulations so specifically that manufacturers know exactly what is required and can adapt to them and, on the other, to not restrict manufacturers unnecessarily in terms of their design options?
- What do we have to require from manufacturers so that, on the one hand, safety and effectiveness are actually demonstrated and, on the other, that this proof (e.g., from clinical investigations) does not unnecessarily endanger patients?
Questions on support and design
- How do we as legislators have to position ourselves in terms of personnel and content?
- What recommendations should we make to authorities and notified bodies to increase their efficiency and effectiveness (e.g., through digitization)?
- How can we divide the tasks in the context of regulatory science between authorities, industry, professional bodies, science, patient (representatives) and others?
- What can we offer to manufacturers to help them? This could include guidelines as well as validated computer models, e.g., of organs.
- Which areas do not have a scientific basis? Where would support be particularly worthwhile?
b) Questions asked by manufacturers
- What is the state of the art (regarding an issue)? How can I identify it?
- What options do I have for demonstrating the safety, performance and effectiveness of my devices? Which of the options are particularly efficient?
- Will the notified bodies and authorities accept these options?
- How can I develop, verify and validate my medical devices quicker using computer models?
- What do I have to do to validate these computer models?
- Which patient population do I have to conduct clinical investigations in? What part of this can I replace with in-silico clinical trials?
Additional information
Read more on computer-based modeling and simulation here and find out how the FDA is using it to encourage quicker development and testing of medical devices.
c) Questions authorities and notified bodies (should) ask themselves
- How do I assess the risks and performance of new
- devices and device classes (e.g., robots)
- technologies (e.g., machine learning)
- methods (e.g., for manufacturing such as 3D printing) and
- materials (e.g., nanoparticles)?
- How do I give myself the biggest possible chance of detecting non-conformities in devices and manufacturers with limited time and human resources?
- In which areas (devices, manufacturers, markets, technologies) should we expect problems in the future? How can we counteract this?
- How can we digitize the authorization process and market surveillance so that we
- detect black sheep faster and more effectively and
- limit our own costs?
- How can I assess how suitable the manufacturers' evidence is for the widest possible range of situations (context of use, patient populations)? How can I spot any gaps?
4. Practicing regulatory science
a) The actors
Regulatory science isn’t just for universities. It is a societal task that requires the cooperation of:
- National and international legislators, authorities
- Notified bodies
- Professional boards such as standards bodies, IMDRF etc.
- Colleges, universities and other research institutions
- Hospitals, clinics and other healthcare facilities
- Patients
- Manufacturers
- Service providers, consulting companies, test laboratories, further education institutes
The Johner Institut has also been actively working for years on:
- Developing guidelines (e.g., on IT security, on artificial intelligence)
- Contributing to the further development of standards
- Developing concepts, data and thought models
- Training young scientists
- Digitizing, and thus increasing the efficiency of, processes such as
- post-market surveillance and
- continuous worldwide research of new and amended regulations
b) The method
The method for regulatory science is science.
Definition: Science
“Science is the totality of human knowledge, insights and experience that is systematically expanded, collected, stored, taught and passed on.”
According to Wikipedia
Scientific work is characterized by the fact that it
- is systematic
- answers questions as completely as possible
- is objective
- is universality valid in the given context and therefore relevant and
- is verifiable
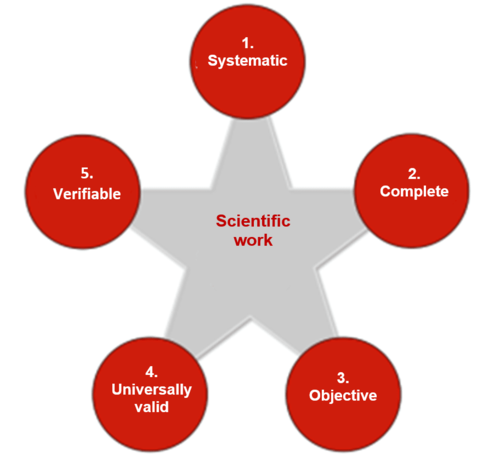
To satisfy these criteria scientists must work systematically and use appropriate methods.
Regulatory science methods include for example:
- Calculations and modeling, e.g., with computer models
- Experiments, including clinical investigations and usability tests
- Analysis of available data, data mining, searching for interdependencies, comparing health systems, devices, methods, materials
- Surveys and observations
- Prototype development
5. Conclusion, summary
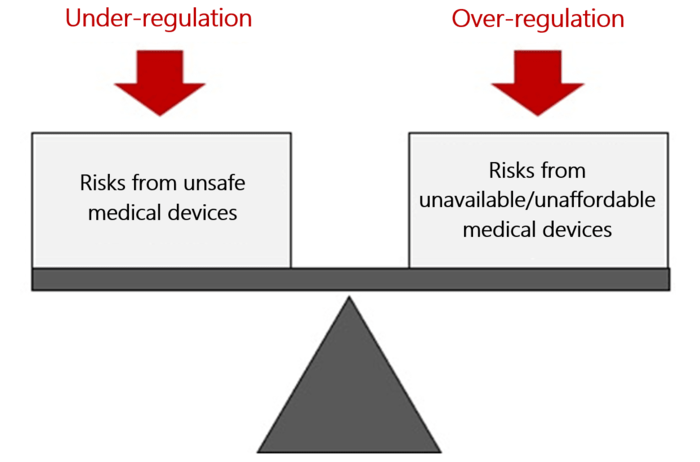
a) The current approach to regulation is regularly too naive
The approach of a lot of regulators, with protecting patients from unsafe devices as the only aim, is not good enough. You could just ban all medical devices and have absolute safety.
But the aim should be to optimize the benefit-risk ratio. This, in turn, requires the benefits and risks to be known and evaluated. And for this evaluation you need data.
Without this data, even well-intentioned legislation degenerates into wild blindfolded “governing”.
Conclusion: Legislators are not meeting the requirements for demonstrating the positive benefit-risk ratio that they demand from manufacturers.
b) It is not just about preventing, it's also about shaping
Professional regulatory science helps
- Encourage innovation
- Support markets
- Ensure that affordable, effective and safe devices are available to the health system
The FDA has long recognized this and is driving regulatory science forward. With over 15,000 employees and a budget of more than USD 3 billion, it has resources that European regulators can only dream of.
c) The regulatory science needs us all, we all need regulatory science
In Germany and many other European countries, the field of regulatory science is largely a desert. The fact that legislators can do better has been demonstrated in the fight against the coronavirus pandemic: Politicians and authorities have involved science/scientists and virologists.
If the approach to battling the pandemic had been the same as the approach to medical device law, the legislator wouldn’t just ignore scientists such as virologists. They’d ignore the entire field of virology.
In order to get from this level more befitting of a third world country to the level of the FDA, e.g., with regard to computer-based modeling, a big effort is required. This can only be achieved together. The Johner Institute is on board.

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.