Medical Devices as Disability Aids: An Additional Source of Revenue?
Health insurance funds reimburse insured people for disability aids, and because disability aids are often also medical devices, this opens up a potential additional source of revenue for some medical device manufacturers.
This article explains which medical device manufacturers can benefit from this “reimbursement” of disability aids and the 5 steps these manufacturers should follow to achieve this.
1st step: find out if the medical device is a disability aid
Section 33 SGB V (German) grants people with statutory health insurance the right to the provision of disability aids. This is a benefit-in-kind.
As a result, a lot of manufacturers are keen to have their device “accepted” as a disability aid by the health insurance funds. To be accepted, the device must meet the definition of a “disability aid”, fulfill the specified objectives of disability aids and meet other criteria.
a) Definition and aims of disability aids
Definition: Disability aid
“Disability aids are items whose use replaces, facilitates, supplements or makes possible an impaired bodily function.”
Source (German)
Disability aids should contribute towards:
- the success of medical treatment (Section 33 SGB V),
- preventing a potential disability (Section 33 SGB V and Section 47 SGB V),
- compensating for a pre-existing disability (Section 33 SGB V and Section 47 SGB V) or
- preventing the need for nursing care (Section 47 SGB V).
There may also be a right to disability aids in the context of preventative healthcare services – for example, to prevent the need for nursing care (Section 11 SGB V, Section 23 SGB V). In addition, disability aids should not be confused with care aids that make the provision of nursing care easier as defined by Section 40 SGB XI.
b) Examples of disability aids
Examples of disability aids include:
- Vision aids (e.g., glasses)
- Hearing aids
- Prostheses
- Orthotics
- Incontinence aids
- Aspirators
- Compression stockings
- Wheelchairs
- Measuring devices such as blood pressure monitors
Products for administering medicinal products into the human body, e.g., syringes, inhalation devices and application aids
Tip
A comprehensive list of disability aids can be found in the disability aids directory. (German)
d) Differentiating between medical devices and disability aids
The medical device and disability aids classes overlap.
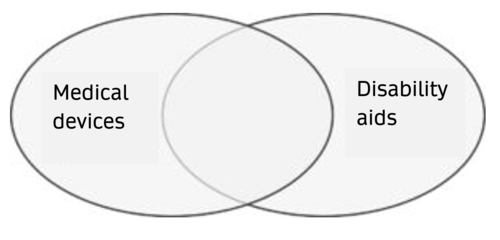
Medical devices that are not disability aids
A lot of medical devices are not disability aids. These include, in particular, products that are (only) intended for use by healthcare professionals. Examples would be X-ray machines, surgical drapes, clinical information systems (German) and laboratory equipment.
Products that are medical devices and disability aids
The definition of the term medical device overlaps with the definition of disability aid. Products that treat or alleviate illness, disability or injury are, by definition, medical devices.
Therefore, wheelchairs, blood pressure monitors, prostheses and aspirators are examples of disability aids that are also medical devices.
Disability aids that are not medical devices
There are products such as toilet seats that do not have an intended purpose (as defined by the manufacturer) that would justify classifying the product as a medical device, but which may still count as a disability aid.
Guide dogs for the blind are also disability aids, but not medical devices.
e) Conclusion
Medical device manufacturers who want to find out whether their product is a disability aid as well as a medical device should check whether it meets the following criteria: The product:
- is in one of the categories in the disability aids directory – if it is not, it is unlikely that the health insurance funds will have to provide the product to insured people,
- is not explicitly mentioned as an excluded disability aid in Section 34 SGB V (German) in the way that all non-prescription, and a lot of prescription, medicinal products are specified as excluded medicinal products,
- is not an a “general item of daily use,”
- is necessary for a specific medical treatment and is not always necessary to achieve the objectives mentioned in a),
- is used by the user and/or their caregivers independently or in self-regulated manner and
- can be carried or taken with them by the person entitled to the product or taken with them if they move house.
Tip
Even if the product is not included in the disability aids directory, the product may still be a disability aid, as the disability aids directory is not exhaustive.
2nd step: work out the item number
a) Why the item number is important
Reason 1: for submitting the right application
To have your medical device included in the disability aids directory published by the Spitzensverbands der gesetzlichen Krankenkassen (GKV-SV) (German), you have to submit an application. In order to find the correct application form, you will need the first seven digits of your future product number for your planned disability aid, the item number.
Reason 2: to identify the quality requirements
You will also need this number to identify the specific quality requirements for the product in the disability aid catalog.
b) How to find the item number
The item number is obtained by working out the:
- The product group
- The site of application
- The subgroup
- The product type
- Your product number. These last three digits are assigned by the GKV-SV
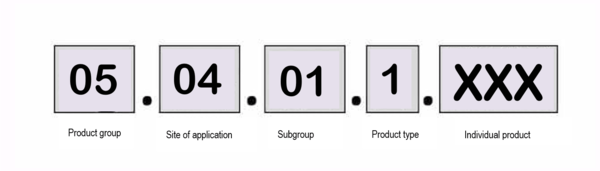
Use the GKV disability aids directory to work out the disability aid number. It provides you with useful details and a description of the product types if you click on the icons shown below.

Details
The details page contains information such as:
- Definition of the product group
- Indication of the product group
- Cross-references to other product groups
- Medical requirements of the subgroups
Product types
Clicking on “Product types” shows a list of all product types broken down by site of application and subgroup.
For example, the product group “electrostimulation devices” contains, among other things, the site of application “nerves/muscles” and below that, among other things, a subgroup “low-frequency electrostimulation devices for pain treatment”.
This subgroup contains two product types, single-channel and multi-channel biphasic pain therapy devices with therapy memory.
3rd step: identify and comply with the (additional) requirements for disability aids
a) Overview of the requirements
Section 139 SGB V (German) describes the requirements to be met by a product before it can be included in the disability aids directory. In particular, the manufacturer must provide evidence of:
- Its functionality
- Its safety
- Its medical benefit (if required)
- The information (in German) that is required for correct and safe use of the product
- Indication-related or use-related quality requirements
- The requirements for service life and reuse
Tip
The disability aids directory defines the indication-related or use-related quality requirements (GKV-SV specific wording). These can be found in the respective product subgroups in the disability aids directory.
The specific quality requirements relate to, for example:
- Indication and use: Functionality, usability, cleaning, compatibility and combination, size, weight, individual adaptation options, etc.
- Service life: Operating life and duration of use, wearing time, etc.
- Reuse, reprocessing, repeated use
NB!
Make sure that you also give the information on these quality aspects in your product’s instructions for use.
b) Proof of the requirements
Functionality and safety requirements
In the case of medical devices, functionality and safety are considered to have already been demonstrated for the CE marking, even if additional testing may be required in borderline cases.
NB!
If non-conformities become apparent during these tests, manufacturers must expect that the umbrella organization will inform the authorities or the notified bodies.
Requirements for demonstrating the medical benefit
Section 139 (5) SGB V does not explicitly state that, for medical devices, the clinical evaluation is enough to demonstrate the medical benefit. But, in practice, the requirements for the clinical evaluation and demonstrating the benefit of a medical device are higher (see, for example, MDR Article 61, MEDDEV 2.7/1 rev. 4) than the requirements for disability aids.
With regard to “clinical investigations”, (German) manufacturers often limit themselves to observational studies and investigations with lower evidence levels.
Usability requirements
The requirements of the MDR and the SGB V are largely the same:
Examples of GKV requirements | MDR requirements in Annex I, Chapter III |
Instructions for use | 23.4(h) |
The intended purpose of the device/indication | 23.4(b) |
Permissible operating conditions/locations of use | 23.4(a) |
Existing risks of use and contraindications | 23.4(g) |
Cleaning instructions/disinfection instructions | 23.4(n) |
Maintenance instructions | 23.4(k) |
Technical data/parameters | 23.4(h, e) * |
Instructions for reuse and the processes required for reuse | 23.4(n) |
Assembly and installation instructions | 23.4(k) |
Information on the material used | 23.4(u) ** |
Table 1: Mapping the requirements for disability aids to the requirements of the MDR.
*: To be listed as performance characteristics or specifications for proper use
**: Limited to certain product groups
Conclusion: Use your instructions for use according to Annex I, Chapter III, Article 23(4) of the MDR as the basis for completing the application for listing as a disability aid and save yourself the work of having to write a new document.
Summary
The following table shows you which documents in your medical device technical documentation and/or clinical evaluation contain the required information:
Evidence required for the application for listing as a disability aid | Primary source in the technical documentation | Link to the clinical evaluation |
Functionality* | Declaration of conformity Where applicable, reports on verification tests/product test reports | The clinical evaluation references test results. The results are incorporated into the safety and performance evaluation. |
Safety* | Declaration of conformity Where applicable, risk management | The clinical evaluation references the risk analysis and identified hazards. It also details other potential hazards and harm as well as their level of severity and probability of occurrence. |
Compliance with quality requirements** | Device description, brochures, test report, etc. | The clinical evaluation references all the functional and technical product specifications from the product description, brochures, etc. and evaluates them. |
Medical benefit | E.g., observational studies according to the GKV-SV requirements | The clinical evaluation proves the clinical benefit and references all the clinical data (e.g., observational studies) on the product. |
Information for proper and safe use in German | Instructions for use | The clinical evaluation references the instructions for use. It checks whether all warnings and safety instructions have been included in the instructions for use. |
Product labeling | Type plate | None |
Table 2: Comparison of the information required for the application and information contained in the technical documentation.
* Functionality and safety are generally considered to have been demonstrated with the declaration of conformity. For novel products, additional documents may be required.
** The quality requirements differ within product groups and product subgroups.
4th step: check that the most common mistakes have been avoided
Manufacturers often submit documents with their application for listing as a disability aid that neither meet the requirements of the applicable standards for medical devices nor the legal requirements.
This applies, in particular, to manufacturers of class I devices, who have problems demonstrating in their technical documentation that their device complies with the general safety and performance requirements.
Factor | Problem | Recommendation |
Interface between instructions for use and risk management | Incomplete listing of product information and warnings for safe use.
This usually relates to the quality requirements for the product and the information in the instructions for use. (Read more after this table). | Check that the GKV’s requirements for the instructions for use have been met. MDR-compliant instructions for use largely meets these requirements – but the legal context of performance (GKV tasks) also has to be taken into account. Read what should be included in the instructions for use and how they should be structured in our article on instructions for use (German) |
Observational studies | Frequently pre-written, unsubstantiated expert opinions from the manufacturer instead of observational studies correctly conducted by independent institutions. | Use product-specific data that demonstrate the clinical benefit and relate to use within the indications. Make sure they meet the GKV-SV requirements. |
Manufacturer-distributor relationship | Incomplete product knowledge, lack of product data | If you are an authorized distributor and are registering a disability aid: request access to the disability aid's technical documentation from the manufacturer. |
Cost reimbursement | Lack of clarity regarding • The location of use • The user group and the associated reimbursement process | Specify all possible locations of use (free market, clinic, hospital, care home, domestic environment, residential environment, etc.) of your product and identify the possible reimbursement processes for your product based on this. |
Table 3: The most common deficiencies in disability aid application documents
5th step: create and submit the application
You should now have all the documents needed to be able to submit your application.
a) Find the right application form
The GKV-SV lists over 118 different application forms that you can download here. Choose the right form for your product based on the seven-digit item number you worked out in a previous step.
b) Complete the application form and attach supporting documents
You already know what you have to demonstrate:
- Functionality
- Medical benefit (if required)
- Safety
- Information for proper and safe use in German
- Compliance with the quality requirements (listed in the GKV)
Submit at least the following documents to the GKV-Spitzenverband:
- Completed application form
- Declaration of conformity used to obtain CE marking
- For class IIa and higher devices: Certificate from the notified body
- Relevant contents of the technical documentation, including
- Device description
- Instructions for use
- Clinical evaluation references (for example, referenced clinical data such as observational studies, etc.)
- Test reports
The MDR already requires these documents and this information in Annexes I and II.
c) Tips for optimizing the application
An incomplete application for disability aid listing delays the inclusion of your medical device in the disability aids directory.
It's not just for you that the documents to be submitted are extensive. The people reviewing your application also need to stay on top of things.
1st tip: check the completeness of the technical documentation
Check our article on the technical documentation to make sure that your technical documentation complies with the requirements of the MDR and, as a result, helps you complete the application for the listing of your disability aid.
2nd tip: use the reference table
Help yourself and the GKV-SV check that your application is complete. To do this, create a table that lists all the quality requirements that apply to your product.
Justify the selection of the quality requirement and enter the references to the supporting documents including the page number.
3rd tip: only submit relevant excerpts
Only submit relevant excerpts from the technical documentation. The reviewer does not need the complete technical documentation. In addition, some documents may be subject to company-specific confidentiality regulations.
Summary
a) Medical device manufacturers have an advantage
Proving functionality, safety and benefit should not be too much of a challenge for medical device manufacturers because the corresponding requirements in the MDR are more detailed and stricter.
Nevertheless, manufacturers should not rely on reviewers waving through their product just because it is a medical device with CE marking.
In the case of class I devices in particular, it often doesn’t become apparent until the application has been submitted that the manufacturer has not sufficiently demonstrated compliance with the general safety and performance requirements or that the technical documentation does not meet the requirements of Annex II of the MDR.
b) It is vital that applications are correct and complete
The Sozialgesetzbuch and case law govern which products the health insurance funds have to make available to the people they insure. And because the health insurance funds are under cost pressures, they will reject incomplete and incorrect applications.
The advice in this article will help you get your product added to the disability aids directory.
c) There are challenges even after inclusion in the disability aids directory
A lot of manufacturers are disappointed with the prices that insurance funds are willing to reimburse. As a result, they are dependent on their products being prescribed at a sufficiently high rate.
It is the manufacturer's job to make sure that the products are actually prescribed. Here too, they have to hold their own against the competition, for example through their unique selling points.
If products are no longer being prescribed often enough, manufacturers could also face the threat of their devices being removed from the disability aids directory.
d) Conclusion
Medical device manufacturers should look at whether they can (also) market their medical devices as disability aids. This review should include a review of the legal requirements and the economic viability of the project.
Additional information
Find out more in podcast episode 2021-06 on disability aids. (German)
Precise technical documentation is an indispensable precondition for marketing both medical devices and disability aids.
Do you still have questions about your technical documentation documents? The Johner Institute will be happy to help you check they are complete. Contact us, for example using the web form, if you would like to know more.
If you have any questions on inclusion in the disability aids directory, Dipl.-Ing. Norbert Kamps, who is an expert in disability aid provision, will be happy to help you.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.